









Serviços Personalizados
Journal
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Costarricense de Ciencias Médicas
versão impressa ISSN 0253-2948
Rev. costarric. cienc. méd vol.19 no.3-4 San José Dez. 1998
Resumen
Se estudiaron dos familias costarricenses con Retinosis Pigmentaria (RP) de herencia autosómica recesiva, con el fin de descartar genes relacionados con la enfermedad. Para esto se efectuó un análisis de ligamiento con marcadores polimórficos por repeticiones en tandem (STRPs). En una familia (C1) los afectados presentan una degeneración de aparición temprana y severa. En la otra familia (P1) la aparición es temprana pero con una degeneración más lenta que en C1. Las diferencias fenotípicas sugieren que se trata de mutaciones diferentes en ambas familias.
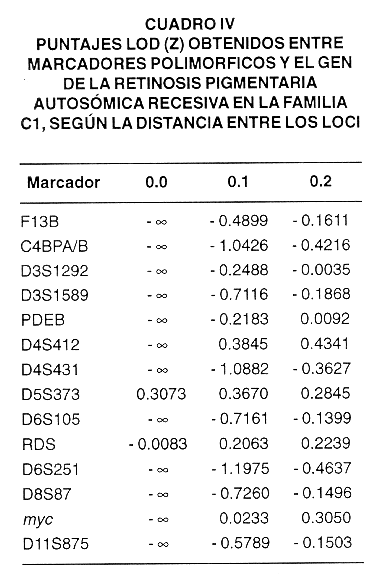
En C1 los siguientes genes quedan descartados (Z (0,0) = ![]() ) : el de herencia autosómica recesiva (RPAR) de la región 1q31-32.1, RPAR por mutación en la fosfodiesterasa B en 4p16.3, RPAR de 6p a 20 cM del gen de la periferina, el de la distrofia macular del cromosoma 6, y del proto-oncogen myc. Por su parte, el puntaje Lod con el marcador RDS no permite descartar con certeza al gen RDS (Z (0,0) =
) : el de herencia autosómica recesiva (RPAR) de la región 1q31-32.1, RPAR por mutación en la fosfodiesterasa B en 4p16.3, RPAR de 6p a 20 cM del gen de la periferina, el de la distrofia macular del cromosoma 6, y del proto-oncogen myc. Por su parte, el puntaje Lod con el marcador RDS no permite descartar con certeza al gen RDS (Z (0,0) = ![]() 0,0083) y se observan puntajes Lod positivos en el caso de los marcadores myc en 8q24.12-q24.13 (Z (0,2) = 0,3050) y periferina/RDS en 6p12 (Z (0,1) = 0.2063), pero no son significativos.
0,0083) y se observan puntajes Lod positivos en el caso de los marcadores myc en 8q24.12-q24.13 (Z (0,2) = 0,3050) y periferina/RDS en 6p12 (Z (0,1) = 0.2063), pero no son significativos.
En P1 se descarta el gen que codifica la rodopsina, el de la fosfodesterasa B, el de RPAR cercano a la periferina, el de la periferina, RPAD del cromosoma 7 y el proto-oncogen myc. Por otra parte, se encontraron valores positivos en marcadores cercanos al gen de la rodopsina (RHO, Z (0.1) = 0.3991), periferina (RDS, Z (0.2) = 0.3390) y Usher 1A del cromosoma 14q (P1, Z (0.09) = 0.7647).
En lo sucesivo deberán descartarse por completo las regiones correspondientes a genes que participan en la transmisión de la señal visual o que forman parte de alguna estructura en la retina. La metodología aquí seguida es útil y factible en Costa Rica para descartar genes implicados en enfermedades hereditarias humanas, y se puede utilizar para conocer los orígenes de otras patologías, realizar un diagnóstico preciso, ofrecer asesoría genética y apoyar el diseño de terapias.
Palabras claves
Genética humana, Enfermedades hereditarias, Diagnóstico molecular, Análisis de ligamiento genético, Retinosis Pigmentaria, Marcadores polimórficos.
Abstract
In order to discard some candidate genes for autosomal recessive Retinitis Pigmentosa (RP), two families with this disease were studied. Linkage analysis was done, using polymorphic markers (STRPs). In one family (C1) affected members present an early onset and severe degeneration of the retina. In the other family (P1) the onset is earlier but the degeneration is slower than in C1. Phenotypic differences indicate that there are different mutations in both families.
The following genes are not responsible for the disease in family C1 (Z (0.0)= ![]() : autosomal recessive RP (arRP) on 1q31-32.1, arRP by mutation in the PDEB gene on 4p16.3, arRP on 6p at 20 cM of peripherin gene, macular dystrophy on chromosome 6 and proto-oncogene myc. On the other hand, the marker RDS´ lod score certainly does not allow discard in a the peripherin gene as responsible (Z(0.1)= - 0.0083), and there are positive lod scores for myc on 8q (Z (0.2)= 0.3050) and peripherin/RDS on 6p12 (Z(0.1)= 0.2063), but they are not significant.
: autosomal recessive RP (arRP) on 1q31-32.1, arRP by mutation in the PDEB gene on 4p16.3, arRP on 6p at 20 cM of peripherin gene, macular dystrophy on chromosome 6 and proto-oncogene myc. On the other hand, the marker RDS´ lod score certainly does not allow discard in a the peripherin gene as responsible (Z(0.1)= - 0.0083), and there are positive lod scores for myc on 8q (Z (0.2)= 0.3050) and peripherin/RDS on 6p12 (Z(0.1)= 0.2063), but they are not significant.
In the case of P1, results suggest that the following genes could be discarded: rodopsin, PDEB, arRP close to peripherhin, peripherin/RDS, adRP on chromosome 7 and myc. Nevertheless positive values were found near regions of rodopsin (RHO, Z(0.1)= 0.3991), peripherin (RDS, Z(0.2)= 0.3390) and Usher 1A on 14q (P1, Z(0.09)= 0.7647).
The next step is to discard the regions with genes implicated in the visual transmission system or in the structure of the retina.
This methodology can be used in Costa Rica for discarding genes implicated in other human hereditary diseases, for ascertaining information on the origen of certain pathologies, in order to get a precise diagnosis, to offer genetic counseling, and to support the design of therapies.
Introducción
La clasificación precisa de enfermedades hereditarias requiere del diagnóstico molecular, esto es, identificar el gen que causa la patología, y preferiblemente, determinar la mutación específica involucrada. De este modo se obtiene información que podría favorecer un mejor pronóstico, un mejor conocimiento del desarrollo de la enfermedad o síndrome y eventualmente, dar luces para la formulación o mejoría del tratamiento. El presente estudio constituye el primer paso en la busqueda de un diagnóstico molecular de la Retinosis Pigmentaria (RP) en familias costarricenses.
La RP es un grupo de enfermedades que se caracterizan por la degeneración progresiva de la retina, ceguera nocturna, pérdida de campo visual y una pigmentación oftalmológica particular (1). La prevalencia de la RP oscila entre 1:3700 y 1:7000 según las poblaciones estudiadas (2-4): en los Estados Unidos los casos se distribuyen en 31% de RP de herencia autosómica recesiva (RPAR), 17% autosómica dominante (RPAD), 10% recesiva ligada al X (RPLX) y 42% a casos aislados (5). El diagnóstico clínico de la RP es sumamente heterogéneo (6), al igual que la genética (Cuadro I). Por ejemplo, en el caso de la RPAR se han encontrado como responsables de la patología mutaciones en el gen de la rodopsina, también implicada en la RPAD, en 3q21-q24 (7), y en el gen de la subunidad B de la fosfodiesterasa en 4p16.3 (8). Además hay loci que se han relacionado por ligamiento con algún tipo de RPAR, como son el del gen del canal catiónico dependiente de GMPc en 4p14-cen, así como el localizado en 14q11, posiblemente debido a una mutación en el gen NRL (9). También se ha encontrado ligamiento entre un tipo de RPAR y el marcador D6S291 en 6p, distinto al gen de la periferina, implicada en la RPAD (10), así como con el marcador F13B en 1q31-q32.1 (11). No obstante, en la mayoría de las familias con RP todavía se desconoce relación con alguna región del genoma (75-80%) (12).
| Cromosoma y gen | Clasificación Clínica | |
| | ||
| 1q31-q32.1 | RP | |
| 1q | Síndrome de Usher II | |
| 3q21-q24 (RHO) | RP | AD, AR |
| 3q21-q25 | Síndrome de Usher III | |
| 3q | RP | |
| 6p | RP | |
| 3 | Sindrome Bardet-Biedl | |
| 4p14-q13 (CNGC) | RP | |
| 4p16.3 (PDEB) | RP | |
| 5q31.2-q24 (PDEA) | RP | |
| 6p (RDS) | Variable | AD, AR |
| 6q14-q16.2 (MCDR1) | Distrofia macular | |
| 7q31-35 | RP | |
| 7p15 | RP | |
| 8p | RP | |
| 11q13 (ROM1) | RP | |
| 11q (miosina VIIA) | Sindrome de Usher I | |
| 11q | Sindrome Bardet-Biedl | |
| 11p15.1-p14 | Sindrome de Usher I | |
| 14q32 | Sindrome de Usher I | |
| 15q22-23 | Sindrome Bardet-Biedl | |
| 16q21 | Sindrome Bardet-Biedl | |
| 17p13 | RP | |
| 19q13.4 | RP | |
| Xp11.23 | RP | |
| Xp21.1 | RP | |
| Mitocondria | Sindrome Kearns-Sayre | Materna |
Es evidente la gran heterogeneidad de la RP, pero se aprecia mejor la complejidad del estudio de este grupo de patologías teniendo en cuenta el hallazgo de una RP provocada por heterogeneidad compuesta. Kajiwara y colaboradores (13) encontraron una familia con un patrón de herencia aparentemente autosómico recesivo, en la que los afectados presentan una mutación en uno de los genes de la periferina/RDS y otra en uno de los genes de la ROM-1 (proteínas que forman un complejo), mientras que los genes correspondientes en los cromosomas homólogos eran normales. Por otra parte, van Soest y colaboradores (11) encontraron una familia con RPAR que, aunque sus miembros tienen ancestros comunes, el origen de la enfermedad se debe aparentemente a dos genes distintos en la misma familia. Los afectados de una parte de la familia tienen el gen ligado a 1q31-32.1, mientras que los afectados de otra parte de la familia no.
El objetivo de este estudio es descartar algunos loci implicados con algún tipo de RPAR, y otros genes relacionados con otros tipos de RP, como responsables de la enfermedad en estas familias. Para esto, se aplica una metodología sencilla con el fin de descartar el gen o los genes que ya han sido localizados en regiones cromosómicas y que podrían ser responsables de enfermedades hereditarias, antes de proceder a un tamizaje genómico completo.
Material y métodos
Clasificación de las familias
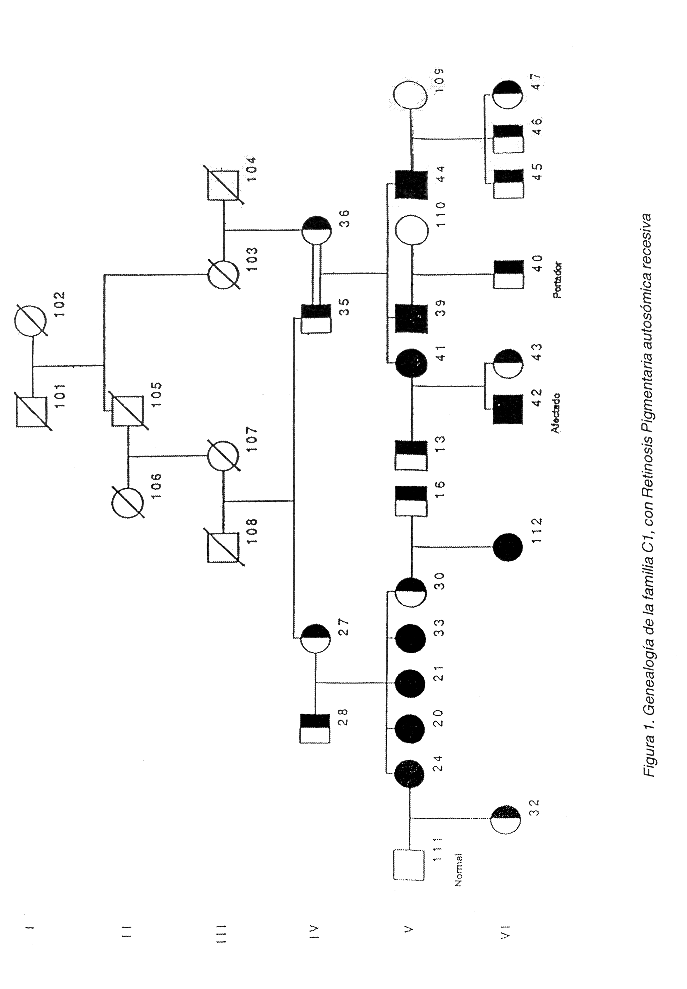
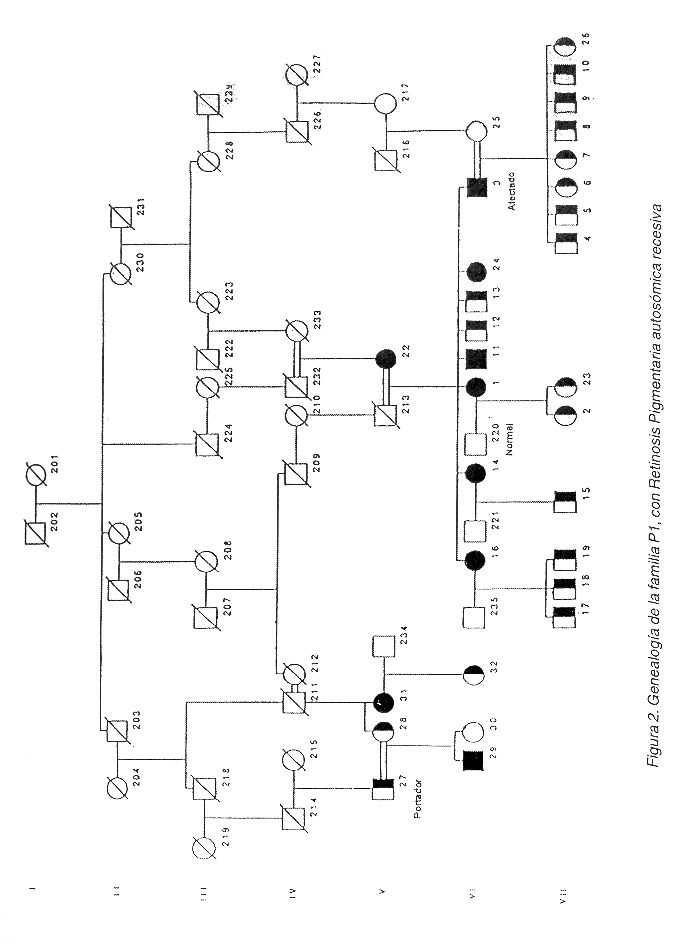
De las 70 familias identificadas por nosotros con RP en Costa Rica (datos no publicados), dos con RPAR fueron estudiadas. La familia "C1" cuenta con 9 afectados vivos y 13 portadores obligatorios vivos (Figura 1), y se analizaron 22 individuos. La familia "P1" tiene 9 afectados y 19 portadores obligatorios vivos (Figura 2), y de ella se analizaron 30 miembros. En esta familia, todos los afectados proceden de uniones consanguíneas, mientras que en la familia C1, no todos los portadores han sido relacionados genealógicamente.
Las familias C1 y P1 se clasificaron en cuanto a su patrón de herencia con la clave de Haim (14). Las genealogías se iniciaron por entrevista y se corroboraron y ampliaron por medio de registros eclesiásticos. Antes de efectuar las entrevistas y análisis se obtuvo el consentimiento informado de los participantes en la investigación.
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
* No se ha relacionado con RP
** Marcador en la misma región pero no se cuenta con la distancia exacta entre el marcador y el gen.
*** Sin número en OMIM por ser loci sujetos a confirmación.
Diagnóstico clínico
A los pacientes y controles se les realizaron estudios de agudeza visual, electrorretinografía (ERG), angiografía y de campo visual en el Instituto de Cirugía Ocular. El ERG se hizo en ambos ojos bajo sedación con hidrato de cloral, con un equipo TOMEY PE-400; se hizo un ERG flash con midriasis y adaptado a la oscuridad por 45 minutos, y se midió la amplitud y tiempo implícito para las ondas A y B (visión escotópica). También se efectuó un ERG flash adaptado a la luz por 5 minutos, y luego se hizo con flash intermitente con frecuencia de 2 Hz/s y se midieron la amplitud (P100) y la latencia (P100 y P70). La angiografía en ambos ojos se realizó con un equipo Top-Con (FD-32 y TRC-50 FT) añadiendo fluoresceína intravenosa, y el estudio de campo visual se realizó con la presentación automática de señales en todo el perímetro y con indicación subjetiva en el momento en que se observaba la señal (Equipo Tomey automático, prueba central 30-2).
Aislamiento, condiciones de pCR, iniciadores utilizados
Se aisló ADN de sangre periférica por el método de fenol-cloroformo (15), se prepararon soluciones de ADN en agua a 0.008 ug/uL y se realizó la Reacción en Cadena de la Polimerasa (PCR). Para esto se utilizaron iniciadores para marcadores polimórficos microsatelíticos (16), reportados en la literatura como ligados a los genes de interés o en regiones cercanas a genes candidatos. Uno de los iniciadores se marcó con 32P (gamma-GTP). Luego se realizó la PCR con las siguientes concentraciones finales: 1X de buffer de reacción (10 mM Tris-HCl, 50 mM KCl), MgCl2 1 mM, cada dNTP a 200 µM, ambos iniciadores a 0.3 µM, 0.33 U de Taq Polimerasa y 28 ng de ADN. El perfil de amplificación fue el siguiente: 94oC (3:00), nueve ciclos de 94oC (0:45), 55oC (1:20) y 72oC (0:45), dieciseis ciclos de 90oC (0.45), 55oC (1:20) y 72oC (0:45), y a 72oC (3:00) para finalizar. El producto de las PCR se corrió en geles de poliacrilamida al 6%, a 55 watts (poder constante), durante 3 a 5 horas. La detección del producto de amplificación se realizó por exposición del gel a una película de Rayos X (Agfa Curix, 100 NIF) por 24 horas.
Análisis de ligamiento
Los marcadores utilizados, su localización y el gen al que están ligados se muestran en el Cuadro II.
Se leyeron las radiografías y se verificó la herencia mendeliana con el programa LINKAGE INTERFASE (17). Luego se utilizó el programa LINKAGE (18) y se observó si había ligamiento entre dos puntos ("Two point analysis") con MLINK y se obtuvo el puntaje Lod como medida de ligamiento (19). El puntaje Lod es una medida de la probabilidad de que dos sitios del genoma (el correspondiente a un marcador y el del gen buscado) estén cerca, contra la probabilidad de que no lo estén. Si dos loci están cerca, la frecuencia de recombinación en meiosis va a ser baja (menor a 0.5), mientras que si dos loci están lejos, la frecuencia de recombinantes será muy cercana a 0.5 (50% de probabilidades de que segreguen juntos, por azar). El puntaje Lod es igual al logaritmo base 10 de la probabilidad de la ocurrencia de los resultados observados en una familia a una fracción de recombinación dada, entre la probabilidad de la ocurrencia de estos resultados por simple azar. Se utiliza el logaritmo porque esto facilita la adición de puntajes Lod encontrados en distintas familias. A saber,
Por convención se acepta que dos loci están ligados si el puntaje Lod es igual o superior a +3 (probabilidad de 1000:1 de que esten ligados) y se descarta ligamiento si el puntaje es igual o menor a –2.
Además, se determinó la capacidad que tienen las familias estudiadas de permitir que se encuentre ligamiento en ellas, para lo cual se crearon segregaciones de cuatro alelos en las familias, uno de los cuales ligado con recombinación de 0.0.
Por desconocerse las frecuencias alélicas en Costa Rica de los marcadores utilizados, se asumió que todas eran iguales para los diferentes alelos, como se suele realizar en estos casos (20). La frecuencia del alelo mutado del gen causante de la RP en cada familia se calculó asumiendo una incidencia de 1:3500 debido a ese gen (21), en cuyo caso la frecuencia génica sería de q = 0.016903.
Resultados
Patrón de herencia y diagnóstico clínico
En ambas familias la herencia es claramente autosómica recesiva de expresión temprana, aunque la degeneración tiene distintas características en C1 y P1.
| | | | | | |
| | | | | | |
| | | | | | |
El fenotipo de la RP en la familia C1 se caracteriza por una respuesta electro-fisiológica nula de bastones, con degeneración variable a nivel de conos (temprana o tardía). Los individuos afectados presentan un moteado difuso temprano en la mácula, que en algunos se presenta muy alterada, con atrofia geográfica. Presentan atenuación vascular, proliferación de pigmento tardía, nistagmus temprano, epitelio pigmentario atrofiado y pérdida de la visión periférica típica de RP.
Por su parte, la familia P1 presenta una respuesta electrofisiológica escotópica nula, pero la respuesta fotópica se agrava tardíamente. Se presenta atrofia en la mácula, atenuación vascular, proliferación de pigmento, pero no nistagmus. El epitelio pigmentario se encuentra atrofiado y se da pérdida de la visión periférica.
Análisis de ligamento
En el Cuadro III se muestra el puntaje Lod de un marcador teórico ligado con el alelo afectado, mientras que en los Cuadros IV y V se presentan los puntajes Lod obtenidos en cada una de las familias. Como puede observarse, ambas familias tienen poder suficiente para que un marcador ligado a la RPAR que las afecta demuestre la existencia de ligamiento (C1, Z (0.0) = 4.59; P1, Z (0.0) = 5.25). Un puntaje Lod de Z (0.0) = ![]() , al tratarse de marcadores polimórificos ligados, permite considerar muy improbable que sea el causante de la patología, al es el caso en la familia C1, con los genes de la RPAR de la región 1q31-32.1 (F13B), RPAR por mutación en la fosfodiesterasa B en 4p16.3 (PDEB), RPAR de 6p a 20 cM del gen de la periferina (D6S105), distrofia macular MCDR1 (D6S251) y del proto-oncogen myc. Por su parte, el puntaje Lod con el marcador RDS no permite descartar con certeza al gen RDS (Z (0.0) = - 0.0083), y se observan puntajes Lod positivos en el caso de los marcadores myc en 8q24.12-q24.13 (Z (0,2) = 0.3050) y periferina/RDS en 6p12 (Z (0.1) = 0.2063), pero éstos no son significativos.
, al tratarse de marcadores polimórificos ligados, permite considerar muy improbable que sea el causante de la patología, al es el caso en la familia C1, con los genes de la RPAR de la región 1q31-32.1 (F13B), RPAR por mutación en la fosfodiesterasa B en 4p16.3 (PDEB), RPAR de 6p a 20 cM del gen de la periferina (D6S105), distrofia macular MCDR1 (D6S251) y del proto-oncogen myc. Por su parte, el puntaje Lod con el marcador RDS no permite descartar con certeza al gen RDS (Z (0.0) = - 0.0083), y se observan puntajes Lod positivos en el caso de los marcadores myc en 8q24.12-q24.13 (Z (0,2) = 0.3050) y periferina/RDS en 6p12 (Z (0.1) = 0.2063), pero éstos no son significativos.
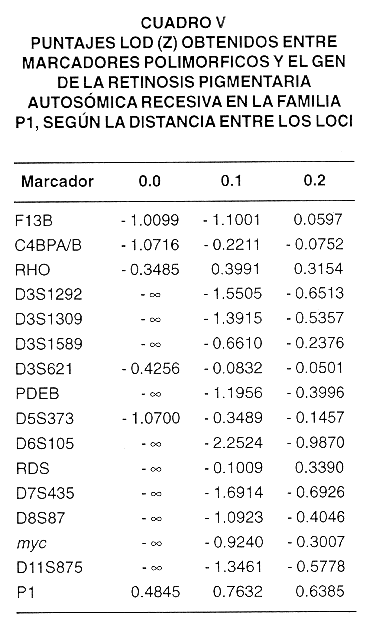
En cuanto a la familia P1, se descartan los siguientes genes como causantes de la RPAR, al obtenerse valores de Z (0.0) =
Ahora bien, hay que tener en cuenta que todos los resultados anteriores corresponden a puntajes Lod obtenidos asumiendo igualdad en la frecuencia de los alelos del marcador, lo cual introduce la probabilidad de error. Sin dejar esto de lado, se puede afirmar que el análisis de los marcadores polimórficos no ha encontrado ninguno ligado a la RPAR en las familias C1 y P1, sino que al contrario, en la mayoría de los casos se han descartado los genes probados.
Discusión
La reconstrucción genealógica de la familia P1 que permitió el establecimiento de nexos consanguíneos como responsables de la enfermedad en todos los afectados, hace pensar que en esta familia una sola mutación es la causante de la patología en todos los afectados. Esto podría facilitar el análisis de ligamiento genético. Aún así no se debe descartar a priori que el problema se deba a la interacción de dos genes en las familias (heterogeneidad compuesta) como ha pasado en otros casos en RP (12). Por su parte, la familia C1 presenta el inconveniente de que no se estableció relación genealógica entre el tronco familiar y algunos portadores obligatorios, por lo que queda una mayor probabilidad de que la heterogeneidad compuesta sea responsable del padecimiento.
Los marcadores utilizados (STRPs) tienen la ventaja de ser muy polimórficos y de permitir un análisis más rápido que si se analizara cada uno de los exones de cada gen, en el caso de contarse con familias que permitan este tipo de análisis. Los puntajes Lod obtenidos para los diversos marcadores ligados a los genes en estudio dan evidencia de que en la familia C1 no hay ligamiento con los genes RP12 (1q31-32.1), PDEB (4p16.3), RP en 6p (a 20 cM de RDS), MCDR1 (6q14-q16.2) y myc (8q24), y quedan por descartarse el gen RDS y regiones cercanas a myc. Por su parte, en la familia P1 se encontró evidencia de que los siguientes genes no causan la RP: RHO, PDEB, RP a 20 cM de RDS, RDS, RP9 y myc. En los casos restantes, en ambas familias se deben probar más marcadores ligados para obtener resultados concluyentes. En ninguna de las dos familias fue posible descartar los genes RP1 y USH1C porque los marcadores ligados (D8S87 y D11S875 respectivamente) no presentaron los puntajes Lod requeridos, si se toma en cuenta la distancia de estos marcadores con respecto al gen (Cuadro II).
Se probó el proto-oncogen myc como posible gen candidato debido a que la proteína codificada por el gen myc participa tanto en el desarrollo del cáncer, como en la apoptosis, mecanismo por el que mueren las células de la retina cuando hay RP. Es predecible que de haberse heredado ese gen con una mutación que predisponga a la apoptosis la muerte celular se daría en estadios tempranos y en diversos tejidos, no sólo en la retina. Sin embargo, se probó porque podría darse el caso de que por los niveles de síntesis proteica en la retina fueran diferentes, y que por tanto, las consecuencias negativas de la mutación se pudieran dar particularmente en ese tejido.
Para continuar con la búsqueda del gen o genes responsables de la RPAR en estas familias y antes de continuar con el mapeo del genoma, deben descartarse los restantes loci que se han relacionado con RPAR y RPAD, así como a otros síndromes con RP. Luego se deben investigar otros genes que codifican proteínas que participan en la transducción de la señal visual o que juegan un papel estructural en células de la retina. Al analizarse los datos se deberá tener en cuenta la posibilidad de que existan dos loci que estén involucrados en la RP en la misma familia. Si estos estudios culminan con el hallazgo de los genes responsables, se estará avanzando en la comprensión de la fisiopatología y se establecerá la posibilidad de un diagnóstico molecular.
En el caso de la RP de estas familias, se ha seguido la metodología habitual de descartar los loci anteriormente relacionados con la patología, para luego continuar con el tamizaje genómico. Cuando se conocen los genes candidatos, se pueden utilizar técnicas directas de búsqueda de mutaciones (p.e. polimorfismos en la conformacion de ADN banda simple, SSCP). No obstante, si se cuenta con familias grandes, con un buen número de afectados, la utilización de marcadores polimórficos y el análisis de ligamiento permite un estudio relativamente barato y rápido. Los pasos seguidos en éste y en múltiples estudios de enfermedades hereditarias, se sintetizan a continuación:
1. Construir la genealogía de la familia afectada.
2. Realizar el diagnóstico clínico más preciso posible de los afectados, portadores obligatorios y controles sanos; de esta manera se escogerán en primer lugar los marcadores más probables.
3. Extraer ADN de los individuos.
4. Realizar pruebas con PCR y los marcadores candidatos, y observar los resultados por medio de electroforesis.
5. Se realiza un análisis de ligamiento, y se descarta o se muestra evidencia del gen como responsable de la patología de la familia.
Después de estos pasos, en caso de no encontrarse ligamiento, se deberá continuar con mapeo genético en sitios del genoma aún no descritos como responsables de la enfermedad.
Agradecimientos
Este estudio es resultado del proyecto # 742-93-324 financiado por la Vicerrectoría de Investigación de la Universidad de Costa Rica.
Agradecemos a Pedro León y a Sandra Silva del Centro de Investigación en Biología Celular y Molecular de la Universidad de Costa Rica, por sus valiosos aportes. A Jorge Azofeifa por sus comentarios. Al señor Alvaro Mendieta y a la Fundación Costarricense de Retinosis Pigmentaria, y al señor Carlos Gamboa y familia, por el importante apoyo económico, necesario por el costo de los estudios clínicos. También agradecemos a Gerardo Jiménez por la ayuda en el trabajo de campo.
Referencias
1. Bunker C, Berson E, Bromley W, Haves R, Roderick T. Prevalence of retinitis pigmentosa in Maine. Am J Ophtalmol 1984; 97: 357- 365. [ Links ]
2. Boughman J, Conneally M, Nance W. Population genetic studies of retintitis pigmentosa. Am J Hum Genet 1980; 32: 223-235. [ Links ]
3. Bundey S, Crews J. A study of retinitis pigmentosa in the city of Birmingham: clinical and genetic heterogeneity. J Med Genet 1984; 21: 421-428. [ Links ]
4. Amman F, Klein D, Franceschetti A. Genetic and epidemiological investigations on pigmentary degeneration of retina and allied disorders in Switzerland. J Neurol Sci 1965; 2: 183-196. [ Links ]
5. Heckenlively J. The diagnosis and classification of retinitis pigmentosa. En: Retinitis Pigmentosa. Philadelphia: JB Lippincott; 1988: 15-24. [ Links ]
6. Farber D, Heckenlively J, Sparkes R, Bateman B. Molecular genetics of retinitis pigmenosa. West J Med 1991; 155: 388-399. [ Links ]
7. Kumaramanickavel G, Maw M, Denton M, John S, Srisailapathy C, Orth U, Oehlmann R, Gal A. Missense rhodopsin mutation in a family with recessive retinitis pigmentosa. Nat Genet 1994; 8: 10-11. [ Links ]
8. McLaughlin M, Berson E, Dryja T. Recessive mutations in the gene encoding the B-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet 1993; 4: 130-134. [ Links ]
9. Bruford E, Mansfield D, Teague P, Barber A, Fossarello M, Wright A. Genetic linkage in autosomal recessive retinitis pigmentosa. Am J Hum Genet 1994; 55 : A181. [ Links ]
10. Knowles J, Shugart Y, Banerjee P, Gilliam C, Lewis C, Jacobson S, Ott J. Identification of a locus, distinct from RDS-peripherin, for autosomal recessive retinitis pigmentosa on chromosome 6p. Hum Molec Genet 1994; 3: 1401-1403. [ Links ]
11. van Soest S, van der Born L, Gal A, Farrar J, Bleeker L, Westerveld A, Humphries P, Sandkuijl L, Bergen A. Assignmento of a gene for autosomal recessive retinitis pigmentosa (RP12) to chromosome 1q31-q32.1 in an inbred and genetically heterogeneous disease population. Genomics 1994; 22: 499-504. [ Links ]
12. Kajiwara K, Berson E, Dryja T. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994; 264: 1604-1608. [ Links ]
13. Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Molec Genet 1995; 4: 1739-1743. [ Links ]
14. Haim M. Retinitis pigmentosa: problems associated with genetic classification. Clin Genet 1993; 44: 62-70. [ Links ]
15. Madisen L, Hoar D, Holroyd CD, Crsip M, Hodes ME. DNA Banking: the effects of storage of blood and isolated DNA on the integrity of DNA. Am J Med Genet 1987; 27: 379-390. [ Links ]
16. Weissenbach J, Gyapay G, Dib C, Vignal A, Morissette J, Millasseau P, Vaysseix G, Lanthrop M. A second generation linkage map of the human genome. Nature 1992; 359: 794-801. [ Links ]
17. Nichols B. Linkage interphase for Macintosh 3.1, Iowa: Molecular Ophthalmology Lab. University of Iowa, 1994. [ Links ]
18. Lathrop G, Lalouel J, Julier C. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 1984; 81: 3443-3446. [ Links ]
19. Morton N. Sequential tests for the detection of linkage. Am J Hum Genet 1955; 7: 277-318. [ Links ]
20. Terwilliger JD, Ott J. Handbook of human genetic linkage, 1 ed., Baltimore, Maryland: The Johns Hopkins University Press, 1994; 307 p. [ Links ]
21. Boughman J, Caldwell R. Genetic an clinical characterization of a survey population with retinitis pigmentosa. En: Cotlier E, Maumenee I, Berman E (eds.) Clinical, stuctural and biochemical advances in hereditary eye disorders: New York: Alan R. Liss; 1982: 147-166. [ Links ]
* Instituto de Investigaciones en Salud y Escuela de Biología. Universidad de Costa Rica. San José, Costa Rica.
E-mail: aleal@humgenet.uni-erlangen.de
{kind=link}
{kind=link}