











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
Permalink
Introduction
Mangrove forests are important keystone of coastal ecosystems and are located along tropical and subtropical areas, covering up to 75 % of the world's coastline. They are productive ecosystems with a unique combination of land and sea, which harbor numerous species of plants and animals (Zhu et al., 2022). Among mangrove plant species, Rhizophora mangle (red mangrove) typically grows in the intertidal regions of the sheltered tropical and subtropical coasts (Ellison et al., 2015). The species R. mangle is native to the Atlantic coast from Florida to Southern Brazil, and in Western Africa, from Senegal to Angola (Ellison et al., 2015), and typically dominates the zone proximal to open water. Such environmental patterns are also observed in Punta Galeta, Panama, where red mangroves stand 10 m from the water limit and extend up to 180 m from the coast's edge (Sousa & Mitchell, 1999). It grows from a shrub to a small tree size, as high as 1-8 m in the Caribbean, and can be reproductively mature at < 1 m (Ellison et al., 2015). This species has adapted their anatomy and developed physiological strategies to survive to the constant changes, such as salinity, nutrients, temperature, and frequent tropical storms, of their environment (DeYoe et al., 2020). Amongst the structures developed by R. mangle to grow and handle harsh conditions, include thickened leaves with secretory glands, flexible stems, and extensive network of roots or rhizophores (DeYoe et al., 2020).
The roots can create a variety of microhabitats suitable for colonization by a range of microorganisms (Gomes et al., 2010; Gomes et al., 2014; Hamzah et al., 2018; Zhang et al., 2017). Culture independent studies conducted on the roots of R. apiculata from China showed that Proteobacteria and Bacteroidetes were the dominant phyla (Zhang et al., 2017). Similar findings were reported in the roots of R. mangle from Brazil, where Proteobacteria and Bacteroidetes also dominated (Gomes et al., 2010). Both studies have in common that within the Proteobacteria, Gammaproteobacteria, and Deltaproteobacteria were the most prevalent and abundant bacterial classes across samples This might suggest the Rhizophora's roots harbor specific bacterial groups, regardless of the geography and the different species of Rhizophora.
On the other hand, culture-dependent studies conducted in R. mangle from Brazil showed low diversity among two different geographical locations with Bacillus being the most common genus, with highest endoglucanase activity (Sá et al., 2014). Other studies have characterized the phylosphere and sediments of R. mangle, and confirmed low bacterial density (Dias et al., 2012). Unlike the phyllosphere, sediments of R. mangle were more diverse being dominated by Alphaproteobacteria and Firmicutes and with the highest endo- and exoglucanase activity (Soares Júnior et al., 2014). The mangrove's sediments were reported to harbor bacteria from orders Vibrionales, Actinomycetales, and Bacillales with promising enzymatic activities (amylases, proteases, esterases, and lipases) for industrial applications (Dias et al., 2009) and antibiotics (Thatoi et al., 2013). Strains belonging to Vibrionales have shown a variety of enzymatic activities, including amylases, proteases (Dias et al., 2009), lipases, and cellullases (Bibi et al., 2017). Most revisions suggest increased Vibrio densities on waters surrounding coastal ecosystems during the summer, due to temperature increase (Johnson, 2015; Takemura et al., 2014). However, exceptions were found in a study with Brazilian mangrove sediments, where Vibrio strains were more abundant during the winter (Dias et al., 2009). Currently there are only few reports exclusively on Vibrio spp. isolated from mangroves (Rameshkumar & Nair, 2009). Studying culturable Vibrio in these marginal forests is essential, as its diversity and composition will help us shed a light on their ecological role.
This study aimed to 1) examine spatial distribution of culturable bacteria in water and sediments along a transect of red mangroves, 2) evaluate the relationship between bacterial densities and their environmental parameters, and 3) explore the cellulolytic potential of the bacterial isolates. We used both selective culture-dependent DNA sequence methods applied to the isolates, to assess the bacterial abundance and diversity along three sampling points across a transect in Galeta Point mangrove forest. Galeta lies next to an urban area and industrial encroachment, due the proximity of Panama Canal and Colon city.
Materials and methods
Study area and site characteristics: The study was conducted in the mangrove forest of Punta Galeta, Panama, near Punta Galeta-Smithsonian Marine Station (9°24' 29.861'' N & 79°51'39.6'' W). A transect of 180 m in length was drawn, which started at the coast and ended up where the red mangrove mixed with the white mangrove. The 180 m transect was divided into three areas. Each sampling site was separated from the other by 80 -90 m in length. GPS coordinates were registered using a GPS eTrex 10 (Garmin, Kansas, US) (Table 1, SMT1).
Table 1 Correlations of environmental parameters in water samples versus bacterial densities transformed to a logarithmic scale.
| Sites | Environmental Parameters | N | Rho | P |
| 1 (0 m) | Salinity | 10 | -0.535 | 0.11 |
| pH | 10 | -0.078 | 0.83 | |
| Temperature | 10 | -0.013 | 0.97 | |
| 2 (80 m) | Salinity | 10 | 0.042 | 0.91 |
| pH | 10 | 0.177 | 0.62 | |
| Temperature | 10 | 0.729 | 0.01 | |
| 3 (180 m) | Salinity | 10 | 0.727 | 0.01 |
| pH | 10 | -0.674 | 0.03 | |
| Temperature | 10 | 0.358 | 0.31 |
N = number of samples; Rho = Spearman's rank correlation; P = probabilistic value. P ≤ 0.05 is considered significant P-values are in bold.
Field sampling collection and environmental measurements: The collection was held on February 9 and 10, 2016 between 9 am and 11 am. The environmental temperature during sampling was 28.6 º C ± 1.19. A total of 60 samples were analyzed at the three transect sites (30 sediment and 30 water samples, 10 per sampling site). Samples of each source were collected aseptically and placed in sterile bags (sediment) at a depth of 5 cm and in 50 ml falcon tubes (water). Samples were transported at 4 °C to the laboratory, where they were processed. Environmental parameters (pH, temperature, and salinity) measurements were made in triplicate, in situ, from ten sediment and ten water samples using Ohaus pens ST10 (pH) and Ohaus ST20s (Salinity and temperature) (Ohaus, New Jersey, USA).
Isolation and bacterial enumeration: Ten water samples (0.1 ml) and sediment (0.1 g) from each site were serially diluted (101-103) with 0.9 ml of NaCl 0.85 %, and plated (0.1 ml) onto Thiosulfate-Citrate-Bile Salts-Sucrose (TCBS) agar (Himedia, Mumbai, India) for total Vibrio enumeration. The culture plates were incubated at 30 °C for 48 h. Because samples were directly plated to this selective medium, with no enrichment steps, the Colony-Forming Units (CFUs) reported here are likely under-estimates of actual bacterial levels (Blackwell & Oliver, 2008). All yellow and green colonies were counted as presumptive vibrio isolates and reported as CFUs per ml of water and CFU per g of sediment.
Screening in Congo red and Lugol agar overlay method: A total of 360 strains were stored at -80 °C in a cryoprotectant solution of Trypticase Soy Broth (TSB) at 2 % NaCl supplemented with 15 % glycerol. Strains were grown at 30 °C on Trypticase Soy Agar (TSA) (Himedia, Mumbai, India) with 2 % NaCl for 24 h. Agar plugs were cut and plated into Zobel marine agar 2216 (Himedia, Mumbai, India) medium containing 1 % Carboxyl methylcellulose (CMC) (Sigma, Saint Louis, USA). Plates were incubated at 30 °C for 48 h. Strains with cellulolytic activity were detected by the formation of clear zones with orange edges around colonies through the Congo red overlay method (Teather & Wood, 1982). A 10 mL aliquot of Congo red dye (2.5 g/l) was then added to each plate. After 30 min, the solution was discarded, and the cultures were washed with 10 mL of 1 M NaCl. Cellulase production was indicated indirectly by areas of hydrolysis. The halos were measured for subsequent calculation of the enzymatic index (EI) using the expression: IE = diameter of hydrolysis zone/ colony diameter (Florencio et al., 2012). The strains that showed CMC hydrolysis were tested for their capacity to degrade agarose. Strains were grown at 30 °C on TSA with 2 % NaCl for 24 h. Agar plugs were cut and placed into plates onto solid Zobel marine agar 2216 medium. Colonies that formed pits or clearing zones on agar plates were dyed with Lugol's iodine solution (1.0 g iodine crystals and 2.0 g KI) and dissolved in 300 mL distilled water (Hu et al., 2009).
Genomic DNA extractions: Due to phenotypic similarities among isolates, a total of 90 strains were randomly selected for genomic DNA extractions according to a previously described method (Blanco-Abad et al., 2009), from an initial 1 ml of each pure culture. Cultures were grown overnight in Luria Bertani broth supplemented with 2 % NaCl at room temperature, then centrifuged at 10 000 rpm for 2 min. The supernatant was discarded, and the pellet re-suspended in 500 µl of 5 % Chelex-100 (BioRad, California, USA). These suspensions were vortexed for a few seconds and incubated at 56 ºC for 20 min, boiled at 100 °C for 10 min and then placed on ice for 2 min and centrifuged at 13 000 rpm for 5 min The supernatant containing the DNA was transferred to a new tube and stored at -20 °C.
PCR amplification and sequencing of 16S rRNA genes: PCR reactions were performed according to the method by Frank et al. (2008). The bacterial 16S rRNA gene was amplified from the genomic DNA, using universal eubacterial 16S rRNA primers comprised by 27F (5'-AGAGTTTGATCMTGGCTCAG-3') and 1492R (5'- TACGGYTACCTTGTTACGACTT-3') (Frank et al. 2008). PCR reactions were performed in a 50 µl reaction mixture containing 3 µL of DNA as the template, each primer at a concentration of 0.4 µM and the mixture of deoxynucleotides triphosphate at a concentration of 0.8 mM, as well as 1 U of FastStartDNA polymerase (Roche Diagnostics GmbH, Mannheim, Germany) and buffer to 1X (Roche Diagnostics GmbH, Mannheim, Germany). PCR amplifications were performed in a 2720 thermal cycler (Applied Biosystems, California, USA) with an initial denaturation for 5 min at 95 °C, 30 cycles consisting of 10 cycles of denaturation at 95 °C for 45 sec, annealing at 58 °C for 45 sec. (decreasing by 0.6 °C per cycle) and extension step of 72 °C for 1.2 min; and 20 cycles of denaturation at 95 °C for 45 sec., annealing at 54 °C for 45 sec. and extension step of 72 °C for 1.2 min plus a final elongation step of 72 °C for 7 min. The PCR product was confirmed by gel (1.6 %) electrophoresis and visualized under UV light after staining the gel with ethidium bromide (0.5 µg/ml). PCR amplicons were purified and sent for custom Sanger sequencing to Macrogen Inc (Seoul, South Korea).
Data analyses: Bidirectional full-length sequences were visually inspected using Geneious R11 (Biomatters Ltd, Auckland, New Zealand) software. A total of 87 isolates passed QC via analyses with Quantitative Insights into Microbial Ecology (QIIME) 1.91(Caporaso et al. 2010). Sequences were inspected for chimeras with the USEARCH61 hierarchical clustering method (Edgar et al., 2011). Sequences were aligned using the SINA aligner (Pruesse et al., 2012). Sequences were binned into OTUs in QIIME using Mothur (Schloss et al., 2009).
Taxonomy: The taxonomy assignment of the representative OTUs was carried out using the RDP database (Wang et al., 2007) with a confidence threshold of 97 %. An OTU table was created using make_otu_table.py using Biological Observation Matrix (BIOM) (McDonald et al., 2012) with the taxonomic classifications. Taxa summaries were built by modifying the QIIME L6 taxonomy table with the R package reshape2 (Wickham, 2007). Pie charts of water and sediment samples were made with the OTU table using MicrobiomeAnalyst (Dhariwal et al., 2017). Taxonomy bar plots with dendrogram and metadata annotations were built using RStudio (https://cran.r-project.org) with the ggplot2 package (Wickham, 2016). We constructed a dendrogram from the results of hierarchical clustering analyses, using the hclust function (Müllner, 2013). The average-linkage algorithm in the hclust model was applied to cluster the samples based on their source. Clustering data was extracted with the ggdendro package (De Vries & Ripley, 2016). The distance matrix was computed using Bray-Curtis distance using the Vegan package (Oksanen et al., 2020). Finally, dendrogram, annotations, and histograms were arranged into a grid using the cowplot library (Wilke, 2017). Megablast analyses was performed to confirm closest match to the NCBI database to some isolates.
Alpha Diversity: Alpha-diversity analyses was done with option -min_rare_depth (the lower limit of rarefaction depths). Chao1 abundance-based estimator (Chao, 1987) was calculated for the water and sediment samples with the script for alpha diversity and boxplots were prepared using the ggplot2 library.
Beta Diversity: Microbial community structure variations between water and sediment samples were performed with beta diversity analyses via Principal Coordinate Analyses (PCoA) on a Bray-Curtis (BC) distance matrix using the Ordinate function from R Phyloseq package (McMurdie & Holmes, 2013). For this, the OTU table and sample metadata were imported in R Studio and merged into one phyloseq object. The OTU counts were transformed into relative abundance matrix previously stored as an OTU table-class. Ordination point colors were assigned according to each sample type variable and customized using aesthetic functions from ggplot2 package.
Statistical analysis: Bacterial counts were log transformed and the distributions of the counts were determined using Shapiro-Wilkson test (P < 0.05). Spearman correlations (Rho) were calculated to evaluate the relationships between the transformed bacterial densities on a logarithmic scale (CFU per mL of water and CFU per g of sediment) and environmental parameters. The significance of the correlation was declared when P ≤ 0.05. All the univariate analyses were carried out in R studio and plots were generated using ggplot2. Significant differences of alpha diversity were calculated using a non-parametric two-sample t-test using 999 Monte-Carlo permutations using the QIIME script compare_alpha_diversity.py using the collated alpha diversity file resulting from the alpha rarefaction analyses. The comparison was done between water and sediments samples, created via the input category passed via ''-c'' on the mapping file. Permutational Multivariate Analysis of Variance (PERMANOVA) was the significance test applied to the beta diversity analyses using the vegan package. This PERMANOVA test is determined through permutations and provides strength and statistical significance on sample groupings using a Bray-Curtis distance matrix as the primary input.
Results
Measurements of environmental parameters by site: Measurements of environmental parameters indicated an increase in salt as measurements were made from the coast to the mangrove forest (varied from 30.85 to 33.68 ppt) (SMT1). Regarding the pH of the water samples, they indicated that the most alkaline point is site 2 (6.99) with a very low pH variation between sites 2 and 3. Water temperature measurements did not vary drastically along the transect. Measurements of environmental parameters in the sediment were fairly constant throughout the transect (SMT2). Site 1 was the coldest because is always flooded. No large variations were observed in the temperature measurements between sites 2 and 3.
Spatial distribution of bacterial densities: All colonies were counted, and bacterial populations were expressed as Log CFUg-1 of fresh sediment and Log CFU mL-1. The highest bacterial density was found in the sediment samples from site 1 (1.53 Log CFU g-1). The lowest bacterial density was given in water samples from site 1 (0.32 Log CFU g-1). Furthermore, for sediment samples, site 2 varied very little in bacterial density compared with site 3 that showed a density of sediment samples. A comparison of bacterial load was made between CFUs from bacteria isolated from water and sediment samples (Fig. 1).
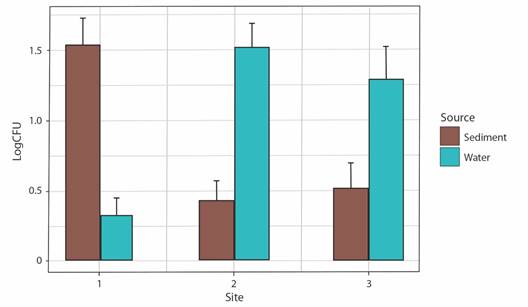
Fig. 1 Distribution of bacterial densities. CFU g-1 (Sediment) or CFU ml-1(Water) across samples and sites along transects.
Relationship between environmental parameters and Log CFU ml-1 or g-1: The relationship between the bacterial densities and the temperature of the water samples indicated a positive correlation (P ≤ 0.05) (Table 1) for site 2 (Rho = 0.729, P = 0.01). In site 3, we found a correlation between the bacterial densities and the salinity of the water samples (Rho = 0.727, P = 0.01. In contrast with positive correlations, we found a negative correlation between pH and Log CFU ml-1 of bacterial densities in site 3 (Rho = -0.674, P = 0.03) (Table 1).
Bacterial isolation: For each site and type of sample (water and sediment), 60 strains were isolated randomly resulting in a total of 360 strains, comprising 180 strains for water samples and 180 strains for sediment samples. All strains showed growth on TCBS, 2 % NaCl TSA, and marine agar 2 216 under the same culture conditions.
Genetic diversity of bacterial strains: A total of 87 good-quality sequences from phenotypically distinct isolates, 42 sequences obtained from sediment strains and 45 obtained from water samples were analyzed using QIIME 1.91. These sequences were binned into 4 OTUs (sediment) and 6 OTUs (water) respectively. Beta diversity analyses shown in the PCoA diagram (Fig. 2A), indicate a slight separation of samples according to sample source (sediment and water). The sediment strains of sites 2 and 3 were grouped close to each other sharing less dissimilarities, compared to OTUs from water strains (Fig. 2A), as also shown by Bray-Curtis dissimilarity analyses separated the water sample from site 1 (RW1) from the rest of water samples (RW2 and RW3) (Fig. 2A). The PCoA showed that the sum of both axes explained almost all (99.6 %) the variation. The sample source, from either water or sediment, is a selectively strong force for the bacterial community structure, rather than each of the sampling sites. Boxplots representing the Chao1 index between sample types showed a higher diversity in the water samples (Chao1 = 4.5) compared to sediment samples (Chao1 = 2.87), with no differences in diversity between type samples (P = 0.274) (Fig. 2B). Although the community is represented by a small number of individuals (rarefaction only by 10 sequences). There was a total of 8 clusters (OTUs) from the combined 87 initial sequences (Fig. 3, SMT3). From the 87 selected strains for genetic characterization, the recovery and selectively of TCBS for Vibrionaceae members was higher in sediments samples (Fig. 3A), obtaining 98 % (N = 41) strains belonging to Vibrionaceae and only 2 % (N = 4) of strains were identified as Proteus. Overall, TCBS showed a high selectivity (88.5 %) by inhibiting the growth of heterotrophic bacteria. The pie charts of water taxa (Fig. 3B) indicated that we could recovered 81 % (N = 36) Vibrionanceae members, and 20 % (N = 9) were identified as non-Vibrionaceae members, including members of Proteobacteria phylum: Ferrimonas, Providencia, and Shewanella. We determined which genera changed between two sources and the three sites depicting the relative abundance through bar plots (Fig. 4). The PERMANOVA test showed no differences between the genera present in water and sediments samples (P = 0.104). Notably, the first site from the water samples within the highest genera diversity, harboring different taxa Vibrionaceae family. The taxa diversity decreased as it entered mainland for both sample types (water or sediment). The taxonomic resolution of 16S rRNA identified Vibrio genera in water samples exclusively. The dendrogram based on dissimilarities indicated that sediments strains shared less dissimilarities than water strains.
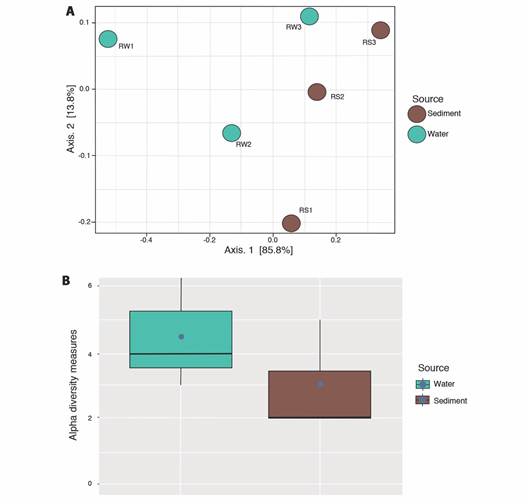
Fig. 2 Alpha and Beta diversity analyses on the 87 isolates. A. Beta diversity comparisons by PCoA. Ordinations of Bray-Curtis dissimilarity between the bacterial strains from two different sources. The color of the spheres indicates the source of the samples. B. Chao1 alpha diversity indexes of strains according to the sampling source. Blue dots indicate the means of the chao1 metric.
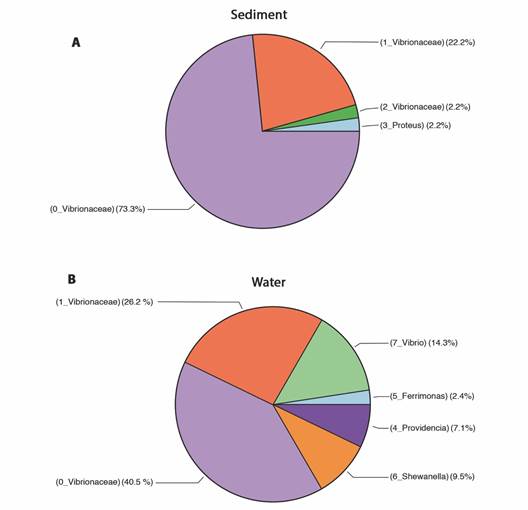
Fig. 3 Pie charts depicting the sample composition among sediment and water samples. A. Relative abundance of the taxa in sediment samples. B. Relative abundance of taxa in water samples. The numbers before each bacterial taxa represent the 8 OTU clusters.
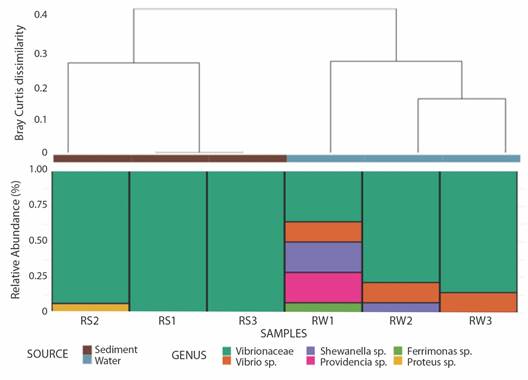
Fig. 4 Taxa summary at the genus levq12el of all bacterial strains. Average-linked clustering (Bray-Curtis) in sediment and water bacterial taxa according to pH, source, and salinity data. Samples derived from sediment are indicated as RS and those from water are indicated as RW.
Screening of cellulose and agar degradation of strains: The overlay method with Congo red was used to evaluate, semi-quantitatively, the ability of the strains to degrade cellulose. All strains (N = 360) were screened for cellulose degradation with 1 % CMC on the marine agar 2 216 in triplicate. This test was based on the observation of growth and measurement of the hydrolysis halo that is used for the calculation of the enzyme index (EI). The halos produced by the cellulose hydrolysis were directly related to the action region of the cellulolytic enzymes, since the dye only remained bound to regions where there likely is b-1, 4-D-glucanohydrolase bonds (Florencio et al., 2012). The absence of color or pale halo around the colonies corresponds to the area of CMC degradation. Strains from sediment that showed CMC hydrolysis on marine agar (4.4 %) were respectively (Sediment site 1: S1-24, S1-31, S1-35 and Sediment site 2: S2-7, S2-8, S2- 9, S2-12, S2-37). Strains isolated from water samples that showed hydrolysis of CMC on marine agar were lower than those of sediment (3.3 %) (W1-5, W1-8, W2-10; W2-30, W3-55, W3-56). The diameter of the halo zone was useful for the selection of strains that can efficiently degrade polysaccharides such as cellulose, xylan and amylose. In addition, the enzymatic index can be used as a simple and rapid methodology to select strains that may have potential to produce enzymes (Florencio et al., 2012). The results of EI (Table 2) obtained for the strains after 2 days of incubation at 30 °C represent the average of measurements for 3 experiments performed independently under the same conditions. The largest enzymatic indexes for cellulose degradation were found in the strains from sediment samples, S1-31 and S2-37 (Table 2). Similar results could be seen in agar degradation with only two strains having agarolytic activity, S1-31 (EI = 4.3) and S2-37 (EI = 3.5). A megablast analyses indicated that the closest match for the strains S1-31 and S2-37 is Vibrio agarivorans.
Table 2 Enzymatic indexes of cellulose degraders and their corresponding halo sizes.
| Strains | Closest match (Megablast) | Mean Halo (mm) | Mean Hydrolisis halo (mm) | EI | SD |
| S1-24 | N.D. | 9 | 2.67 | 0.30 | 0.58 |
| S1-31 | Vibrio agarivorans | 9 | 43.33 | 2.41 | 1.53 |
| S1-35 | N.D. | 9 | 3.67 | 0.41 | 0.58 |
| S2-7 | N.D. | 9 | 4.67 | 0.52 | 0.58 |
| S2-8 | N.D. | 9 | 5.67 | 0.63 | 0.58 |
| S2-9 | N.D. | 9 | 7.67 | 0.85 | 0.58 |
| S2-12 | N.D. | 9 | 8.67 | 0.96 | 0.58 |
| S2-37 | Vibrio agarivorans | 9 | 33.67 | 2.59 | 1.53 |
| W1-5 | N.D. | 9 | 2.00 | 0.22 | 0.00 |
| W1-8 | N.D. | 9 | 1.67 | 0.19 | 0.58 |
| W2-10 | N.D. | 9 | 3.67 | 0.41 | 0.58 |
| W2-30 | N.D. | 9 | 2.67 | 0.30 | 0.58 |
| W3-55 | Vibrio alginolyticus | 9 | 5.67 | 0.63 | 0.58 |
| W3-56 | N.D. | 9 | 3.67 | 0.41 | 0.58 |
EI = enzymatic indexes. N.D. = Not determined; SD=Standard deviation.
Discussion
The bacterial densities of cultured bacteria along a red mangrove transect in Panamá showed a pattern defined according to the sample origin (sediment and water). We observed that in water samples the bacterial densities increased as it entered mainland; the inverse happened in sediment samples. However, the abundance remained steady in sites 2 and 3 for both types of samples. This invariability in Vibrionaceae densities was like a distribution pattern found in sediments of three different sites from Mexico populated by black mangroves (Gonzalez-Acosta et al., 2006). Although our results share a common stability pattern, the abundance of culturable Vibrio spp. in sediments of Mexico was higher (7.4 log CFU g-1) (Gonzalez-Acosta et al., 2006) compared with our bacterial densities in sediments (0.82 Log CFU g-1). A study on Brazilian mangrove sediment bacteria (Dias et al., 2009), showed no recovery of Vibrio spp. from mangroves sediment at a depth of (0-10 cm). Conversely, we could isolate Vibrionaceae members from our sediment samples at a depth of 5 cm. Regarding the water samples, the densities of Vibrio spp. in estuaries from some investigations during the dry season were similar to our strain densities in water (Pfeffer et al., 2003; Turner et al., 2009; Wetz et al., 2008).
Temperature and salinity have been the most exhaustively studied environmental factors associated with Vibrio densities (Johnson, 2015; Takemura et al. 2014). Our results are supported by other studies where positive correlation with temperature (Janelidze et al., 2011; Pfeffer et al., 2003; Wetz et al., 2008) and salinity (Pfeffer et al., 2003; Turner et al., 2009) were observed in estuarine waters, using similar culture techniques. Our correlation analysis suggested that the environmental factors were the driven forces to shape the culturable community in water samples, where we found that alpha diversity was higher compared to the sediment. Site 1 made the highest contribution to the taxa abundance reported in water samples. One possible explanation about this was this site was the closest to the sea and therefore was under the influence of tidal hydrodynamics, which probably led to the selection of different taxa. It is likely that spatial distribution of Vibrionaceae was higher in water samples of the first site due several factors that go beyond the focus of this study, such as: residence time, net growth rate, grazing rates by protists, stress, nutrient availability, and viral lysis rates (Mackey et al., 2017). Overall, the variation of the bacterial communities in the mangrove ecosystem is characterized by periodic flooding of the tides; and environmental factors such as salinity, temperature and nutrient availability are highly variable and result in unique and specific features (Holguin et al., 2006).
In this study, we observed that the sample type (water/sediment) have a high impact in the composition of Vibrionaceae taxa compared to each sampled mangrove habitat. Conversely to our findings, a study conducted in Brazilian mangrove sediments, found that each site of a transect select and influence specific the bacterial communities (Rocha et al., 2016). This same study reported the prevalence of Vibrio in their first collection site as well in the last section of them transect (site 3). Although their transect was longer than ours - 450 m distance- (Rocha et al., 2016), it revealed similar Vibrio levels to those reported here.
According to our genetic characterization at the genus-level, Proteus was only found to be present in one of the sediment samples - a bacterium known to be an indicator of fecal contamination (Drzewiecka, 2016). We also report the presence of Shewanella, Providencia, and Ferrimonas in water samples. Despite the selectivity of TCBS for vibrio isolates, other genera such as Staphylococcus, Flavobacterium, Pseudoalteromonas, and Shewanella can grow on TCBS as well, (Thompson et al., 2004) as we found. Perhaps, these non-Vibrionaceae members have a terrestrial origin as a result of freshwater inputs. The identification using 16s rRNA gene showed a good resolution for water strains at the genus level. This limitation could be circumvented using different phylogenetic and evolutionary methodologies such as Multi-Locus Sequence Typing (MLST), Multiple-Locus Variable-Number Tandem-Repeat Analysis (MLVA), or Pulsed-Field Gel Electrophoresis (PFGE) which allows to distinguish strains with little genetic variation (Lüdeke et al., 2015). Factors that maintain genetics and species diversity act in concert in marine ecosystems (Robinson et al., 2010). In this research, changes in species composition and other levels of diversity (genetic and species diversity) along the study gradient seems to be both related to substrate water exposure. The same pattern has been observed in other benthic intertidal taxa where water exposure, salinity, temperature, habitat complexity and substrate spatial heterogeneity shape the community structure and diversity (Archambault & Bourget, 1996).
In general, there are several studies assessing and predicting the dynamics of Vibrio as these are impacted by recreation, used in animal aquaculture and, therefore, represent a public health issue with consequences for humans and marine animals (Jacobs et al., 2014; Johnson, 2015; Lüdeke et al., 2015; Takemura et al., 2014; Turner et al., 2009; Wetz et al., 2008). However, a few of these exploit the biotechnological capabilities of Vibrio to degraded natural biomass, despite the diverse physiological properties and ubiquity in marine environments. We showed that in our transect, the strains that produced the largest hydrolysis halo on CMC media (S1-31, S2-37) and caused a depression in the agar, were positive for hydrolysis of agar using Lugol. Interestingly, few Vibrio spp. have been reported to possess multi-enzymatic machinery (Dias et al., 2009, Liu et al., 2016), as those explained here from red mangrove sediments. To the best our knowledge, our study reports for first time the presence of V. agarivorans in tropical mangroves. The first and only report of V. agarivorans comes from Mediterranean seawater, with which our two strains matched (Macián et al., 2001). Further physiological characterizations are needed as well as quantifications of the hydrolysis with the test of reducing sugars with DNS (Di-nitro salicylic acid) for these strains. This could shed a light to how these two Vibrio strains adapted to the environmental challenges present in the red mangrove forest of Punta Galeta, being able to degrade 2 different substrates.
Vibrio is a natural inhabitant along the three sites of our mangrove transect and is well known for its high frequency of gene exchange (especially some clades), leading to a very rapid evolution and genomic plasticity (Tagliavia et al., 2019). In this sense, we consider that sediment strains might adapted to different characteristics in this habitat leading to expression of higher enzymatic capabilities than their counterpart water strains. Vibrio species are Gram-negative, curved, rod-shaped bacteria belonging to the class Gammaproteobacteria and are still regarded by most marine microbiologists as the dominant culturable bacteria in the ocean (Pruzzo, et al., 2005). This feature made Vibrio a model to screening enzymes with biotechnological applications because working with the living organisms allows to overcome some obstacles, such as heterologous expression of proteins encoded by environmental DNA in a surrogate host (Tagliavia et al., 2019), generated while performing functional metagenomics to screen for microbial enzymes.
Spatial analysis like this, joined with molecular genotyping, could provide important information to identify microbial hotspots as well as deserts in mangrove transects (Gonzalez et al., 2012). Additionally, correlations with environmental parameters help develop new hypothesis about dispersal limitation (Gonzalez et al., 2012). Although sometimes it is difficult to assign these physiological features to species (Tagliavia et al., 2019). There is much interest on the biotechnological capabilities of mangrove microbes in which the variable environmental conditions may impact microbial structure and the enzymatic properties of the communities.
This study reports the diversity of Vibrio along a red mangrove transect in Panama, indicating a moderate Vibrio load during the time lapse of sampling and along a 180 m transect. The correlations between the Vibrio spp. load and temperature, indicate that this parameter is a strong environmental driver for the detection of Vibrio. Genetic analyses based on the 16S rRNA gene indicate that there is a higher diversity of Vibrio in water samples compared to sediment, although Vibrio strains from the sediment have cellulolytic activity. In summary, our report highlights the isolation of cellulose-degrading Vibrio from the red mangrove niche, and indicates its potential applicability in various industrial processes such as biofuel production.
Ethical statement: the authors declare that they all agree with this publication and made significant contributions; that there is no conflict of interest of any kind; and that we followed all pertinent ethical and legal procedures and requirements. All financial sources are fully and clearly stated in the acknowledgements section. A signed document has been filed in the journal archives.

