











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
La hemoglobina en humanos está compuesta por 4 cadenas de globina (2 cadenas α y 2 cadenas β) y un grupo hemo que transporta el oxígeno. Las cadenas de alfa-globina están codificadas por genes localizados en el cromosoma 16 y los genes que codifican para las cadenas de beta-globina están localizados en el cromosoma 11. En el adulto sano, las hemoglobinas presentes son la HbA (2a/2β) en una concentración de entre el 95% y el 98%; la HbA2 (2a/2δ) en una concentración menor a 3.5% y la Hb Fetal (2a/2γ) en una concentración inferior al 1%.1
Los desórdenes hereditarios de la molécula de hemoglobina están dentro de los desórdenes genéticos clínicamente más comunes, de los cuales existen dos tipos: aquellos en los que las mutaciones interfieren con la cantidad de proteína producida (talasemias) y aquellos que resultan en cambios estructurales de la molécula de hemoglobina y que causan una variante proteica (hemoglobinopatías).1
Las talasemias tipo alfa son enfermedades monogénicas comunes en el ser humano (5% de la población mundial) y ocurren con mayor frecuencia que las beta-talasemias.1 Las alfa-talasemias son heredadas de forma autosómica recesiva y se caracterizan por un déficit en la producción de las cadenas de globina a de la hemoglobina. Esta condición se presenta con mayor frecuencia en áreas tropicales y subtropicales, con una proporción de entre 80-90% de portadores. Las formas clínicas severas de enfermedad son la enfermedad por Hb H y el hidrops fetalis, con ausencia en la producción de tres y cuatro cadenas alfa respectivamente.
Se han identificado más de 100 genotipos de alfa-talasemia ampliamente distribuidos a nivel mundial. La mayor parte de los casos son producidos por defectos delecionales en los genes HBA1 y HBA2.2 La deleción más frecuentemente encontrada es la de tipo -3.7 Kb, que involucra uno de los 4 genes de alfaglobina, y existen formas que involucran dos o más genes, como las deleciones de tipo --MED, --SEA y -FIL.1,2 Con menor frecuencia, las alfa-talasemias son causadas por mutaciones puntuales en los genes de alfa-globina que comprometen predominantemente la expresión del gen HBA2 y raramente en el gen HBA1. Estas variantes no delecionales dan lugar a variantes de hemoglobina, como la Constant Spring, Icaria, Koya Dora, entre otras.1,2
El espectro clínico de las alfa-talasemias es muy variable y su clasificación fenotípica está relacionada con el número de alelos afectados. Un individuo con afectación de un solo alelo se conoce como portador silente (un alelo disfuncional); la presencia de dos alelos no funcionales se caracteriza como rasgo a talasémico (dos alelos disfuncionales), en cuyo caso la persona, si ha heredado un alelo disfuncional de cada progenitor, tendrá una condición homocigota; y si ambos alelos disfuncionales heredados son del mismo progenitor, será de condición heterocigota.2,3 La ausencia de 3 alelos (--/-a) determinará la enfermedad por Hb H, la cual es un tetrámero (β4) de las cadenas de beta globina sobrantes. En caso de que exista una ausencia total de genesa(--/--) surgirá la condición llamada síndrome de hidrops fetalis por Hb Bart.1,2
Los individuos afectados por la condición de rasgo silencioso o rasgo a-talasémico presentan anemia leve en el hemograma, con un volumen corpuscular medio (VCM) menor a 80 fL, una hemoglobina corpuscular media (HCM) menor a 27 pg y un cómputo de glóbulos rojos normal o aumentado para el grado de anemia presente. Los pacientes con las formas más severas, enfermedad por Hb H o hidrops fetalis, presentan niveles de anemia severos.3,4
Desde el punto de vista clínico, en los casos de portador silente y de rasgo alfa-talasémico, el paciente se muestra asintomático con poca o ninguna repercusión clínica, además puede tener un hemograma prácticamente sin alteraciones, con anemia microcítica-hipocrómica de leve a moderada, detectada mediante un hemograma de rutina. En la enfermedad por Hb H, los hallazgos clínicos predominantes son compatibles con anemia hemolítica, presencia de Hb H en la electroforesis de hemoglobina, esplenomegalia acompañada, en algunas ocasiones, de hiperesplenismo e ictericia. Otras complicaciones que se pueden presentar son: infecciones, úlceras en las extremidades, cálculos biliares y episodios de anemia hemolítica desencadenados por cuadros infecciosos o por medicamentos. La presentación clínica más severa de la enfermedad es el síndrome de hidrops fetalis¸ que ocurre cuando hay ausencia absoluta de genes a globina, lo que causa la aparición de tetrámeros de g4 (Hb Bart) que no son funcionales. Los hallazgos clínicos de inicio son: anemia intrauterina, aspecto edematoso al nacer con pronunciada hepatoesplenomegalia, signos de insuficiencia cardiaca y deformidades esqueléticas y cardiacas. Los portadores de este síndrome suelen fallecer en el útero entre las 23-38 semanas de edad gestacional o poco después del nacimiento, por lo que en estos casos el consejo genético en parejas progenitoras es indispensable.3,4
En Costa Rica, a partir de octubre del 2005, se incluye la detección de hemoglobina S, tanto homocigota como heterocigota, en la prueba de tamizaje neonatal, lo que permite detectar a los pacientes drepanocíticos y a aquellos pacientes portadores. En el 2012, se inició un proyecto de secuenciación que faculta el análisis molecular de hemoglobina S y C en la misma muestra del tamizaje neonatal. Para el 2015, se implementó la secuenciación completa de todo el gen beta, lo que posibilitó la detección de talasemias beta; en el 2017, para el gen beta se utilizó una técnica más específica llamada ampliación de sondas dependiente de ligandos múltiples (MLPA, por sus siglas en inglés). Esta prueba ha permitido una detección temprana de hemoglobinopatías S y beta-talasemia en la población costarricense; sin embargo, al gen alfa no es posible analizarlo con la prueba de tamizaje neonatal; por ende, una sospecha médica temprana es fundamental para ampliar estudios en el laboratorio especializado de hematología de Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”.
A la fecha, no se dispone en el país de un estudio descriptivo y epidemiológico de alfa-talasemia en población infantil a nivel Nacional , por lo que surge la necesidad de realizar un examen clínico observacional para describir y analizar las características clínicas y epidemiológicas de los pacientes con alfa-talasemia atendidos durante el periodo del 1° de enero 2018 al 31 de enero 2019 en el Servicio de Hematología del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”, de la Caja Costarricense de Seguro Social (HNN-CCSS).
Métodos
Se realiza un estudio observacional, descriptivo, transversal, con revisión retrospectiva de expedientes de los pacientes referidos al Servicio de Hematología del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”, de la Caja Costarricense del Seguro Social, para ser evaluados ante sospecha de alfa-talasemia, en el periodo comprendido entre enero del 2018 y enero del 2019. El estudio se aprobó por el comité ético científico del HNN-CCSS con el código CEC-HNN-022-2020, de acuerdo con los principios éticos incluidos en la Declaración de Helsinki y de Buenas Prácticas Clínicas.
Muestra: Se utilizaron expedientes clínicos, electrónicos y registros de laboratorio de pacientes referidos al Servicio de Hematología del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”, de la Caja Costarricense del Seguro Social, con el diagnóstico de anemia microcítica-hipocrómica refractaria a la terapéutica con hierro oral. Se incluyó en el estudio a aquellos pacientes con un rango de edad entre los 0 meses hasta 12 años y 11 meses cumplidos al momento del estudio, sin distinción de género ni de etnia, que presentaban microcitosis (VCM<80 fL), hipocromía (HCM < 27 pg) y electroforesis de hemoglobina con patrón de distribución normal AA, con HbA2 en rango normal (2-3.3%) o disminuida, y que además debían ser pacientes atendidos en el Servicio de Hematología del HNN, CCSS, durante el periodo entre enero del 2018 y enero del 2019.
Para el estudio fueron avalados los pacientes con información completa en su expediente, con un hemograma de referencia inicial, electroforesis de hemoglobina y un estudio molecular de alfa-talasemia realizado en el laboratorio de Estudios Especializados e Investigación del hospital. Se excluyó a aquellos que carecían de más de un 30% de las variables por estudiar y que no contaban con un estudio molecular por alfa-talasemia.
Estudio molecular: se utilizó amplificación por PCR e hibridación inversa del ADN genómico en leucocitos de sangre periférica de los pacientes. Dicho análisis molecular incluyó la detección de las deleciones/mutaciones: 3.7, 4.2, 20.5, MED, FIL, SEA, THAI, anti-3.7 triplicación, HbConstant Spring, HbQuonSze, Hb Adana, HbKoya Dora, HbIcara, HbPakse, a2 poli A-1/2, a2-cd142, a1-cd14, a2-init-cd, a2-cd19, a2-IVS1, a2-cd59.
No se realizó estudio de laboratorio o gabinete extra ni metodología intervencionista. Los datos se recolectaron en formulario validado por el Comité de Bioética, el cual aprobó el estudio, y se computaron con Microsoft Excel.
Análisis de datos: se estimaron las frecuencias y porcentajes en las variables cualitativas, la media para las variables cuantitativas y la desviación estándar como medida de dispersión. Se determinó la frecuencia para las variables de sexo, edad categorizada al momento del diagnóstico, provincia de residencia, motivo de referencia, parámetros hematológicos y defectos genéticos encontrados en el análisis molecular. Se estimaron medias y desviaciones estándar para las siguientes variables: conteo de glóbulos rojos al momento del diagnóstico, hemoglobina, VCM y HCM, parámetros de evaluación del metabolismo de hierro y fracciones de hemoglobina evaluadas mediante electroforesis de hemoglobina.
Para la evaluación de la distribución poblacional y según la provincia de residencia, se estimó la prevalencia por cada 100 000 niños menores de 13 años, tomando como denominador la población de este grupo de edad según las proyecciones oficiales del Instituto Nacional de Estadísticas y Censos disponibles en el sitio oficial de la institución para el 2018 (Instituto Nacional De Estadística Y Censos. San José: INEC; 2018. [accesado 04-03-2020]. Disponible en: https://www.inec.cr/sistema-de-consultas). Todos los análisis fueron desarrollados por medio de Stata 15.1 (StataCorp, 2019 Texas, USA).
Resultados
Se incluyó a un total de 60 pacientes. La distribución según sexo fue de 55.0% (33/60) para el sexo masculino y un 45.0% (27/60) para el femenino. De los 60 casos estudiados, se confirmó la enfermedad en 44/60 casos (73%). El grupo etario con mayor frecuencia de alfa-talasemia fue el de 0-5 años, con un 71.6% (43/60) de los casos, seguido del grupo de 6-10 años de edad, con 23.3% (14/60), y por último los mayores de 11 años, con 5% (3/60) de los casos. Al evaluar la distribución de la totalidad de los casos según provincia, la mayor frecuencia estuvo en San José, con 33.3% (20/60), seguido de Limón con 18.3% (11/60) y Puntarenas con 15.0% (9/60). Al realizar el análisis de prevalencia por provincia en el periodo en estudio, se constató que las mayores prevalencias fueron reportadas para la provincia de Limón, con 10.8 casos por cada 100 000 habitantes menores de 13 años; Guanacaste y Puntarenas representaron ambos el segundo lugar con igual número de 8.5 casos por cada 100 000 habitantes respectivamente; San José presentó una prevalencia de 7.1 casos y Heredia, Alajuela y Cartago reportaron prevalencias de 5.2, 3.1 y 2.0 casos por 100 000 habitantes menores de 13 años respectivamente (Figura 1).
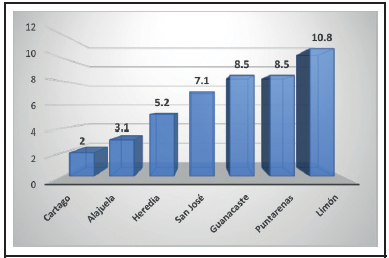
Figura 1: Distribución según provincia de procedencia de pacientes referidos al servicio de Hematología del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera” Caja Costarricense de Seguro Social, para estudio por alfa-talasemia, según tasa de prevalencia por 100.000 mil habitantes menores de 13 años. Enero 2018 - enero 2019. N=60
Al analizar los motivos de referencia, se encontró que la anemia microcítica hipocrómica representó el 36.7% de los casos (22/60), siendo el motivo más frecuente de derivación al Servicio de Hematología en el periodo de estudio; seguido de la anemia con pobre respuesta al tratamiento en el 28.3% de los casos (17/60) y, por último, otros motivos de referencia entre los que se encontraban: anemia en estudio, anemia y trombocitosis, lo que corresponde al 18.3% de la población estudiada. Según los parámetros hematológicos iniciales, la media obtenida para los glóbulos rojos al momento de referencia fue de 5.07 x106/μL (DE 0.69), además de un promedio de hemoglobina de 10.8 g/dL (DE:1.1), VCM con una media de 65.7 fL (DE:7.0) y HCM con una media de 21.9 g/dL (DE:3.5) para la población del estudio (ver Cuadro 1).
Cuadro 1: Parámetros hematológicos y su valor medio en pacientes referidos al servicio de Hematología del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”, Caja Costarricense de Seguro Social, para estudio por alfa-talasemia de enero 2018-enero 2019. N=60
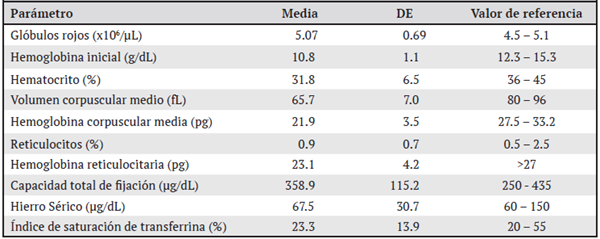
El valor medio de las fracciones de hemoglobina determinadas por electroforesis capilar es: HbA un 96.4% (DE:4.2), HbA2con un 2.4% (DE:0.5) y HbF con una media de 0.3% (DE:0.6) para los sujetos del estudio. En 3 de los 60 pacientes estudiados, se detectó Hb H en la electroforesis. Para el conteo de glóbulos rojos totales, se obtuvo que en el 76.7% (46/60) fue hasta un máximo de 5.5 (106/μL); el rango de hemoglobina oscilaba entre 10 a 10.9 g/dL (23/60) en el 38.3% de los casos. En relación con el VCM, el 90.0% de los pacientes presentó un valor menor o igual a 75 fL y un HCM menor o igual a 24 pg/dL en el 75.0% de los casos, lo que determina microcitosis e hipocromía respectivamente.
En los pacientes con sospecha inicial de alfatalasemia, se descartó la anemia por deficiencia de hierro mediante la obtención de parámetros de evaluación del metabolismo del hierro. Se consideró como anemia carencial aquellos casos que tuvieran valores de hierro sérico menores de 50 μg/dL, capacidad total de fijación superior a 255 μg/dL e índice de saturación de transferrina inferior al 20%, como lo establece la literatura.5
Se identificó que en el 21.7% (13/60) de los casos compatibles con alfa-talasemia, se presentaron a la vez parámetros sugestivos de anemia ferropénica.
De los 60 pacientes reclutados, el 73.3% (44/60) demostró la condición de alfa-talasemia confirmada mediante el estudio molecular y en un 26.6% (16/60) de los casos no se logró establecer el diagnóstico de alfa-talasemia, al menos con las mutaciones incluidas en el análisis molecular.
La edad media de diagnóstico de los pacientes confirmados con alfa-talasemia fue de 4.9 años, con un rango de edad de 0 meses a 12.0 años. La distribución según sexo fue 52.3% (23/44) para el sexo femenino y 47.8% (21/44) para el masculino.
En cuanto al antecedente de familiares con hemoglobinopatía, se reportó la presencia en la historia clínica de este hallazgo en un 36.3% (16/44) de los niños confirmados por alfa-talasemia.
En los sujetos del estudio, el tipo de defecto genético más frecuente fue la deleción 3,7 Kb con un 77.2% (34/44) de los casos, seguido de la deleción SEA en el 15.9% (7/44) (Cuadro 2).
Cuadro 2: Distribución de pacientes diagnosticados con alfa-talasemia según genotipo y cigocidad en la población atendida en el Servicio de Hematología del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”, Caja Costarricense de Seguro Social. Enero 2018 - enero 2019. N=44
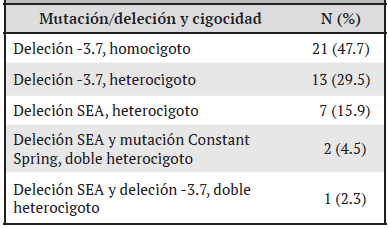
De los 44 pacientes con defectos moleculares confirmados para el gen de alfa-globina, se demostró que la distribución según rasgo de cigocidad fue equitativa tanto para los heterocigotos como para los homocigotos, contemplando un 47.7% (21/44) de los casos respectivamente; además, se encontró que en el 6.8% (3/44) la condición fue de doble heterocigoto.
Se halló que el fenotipo más frecuente de los pacientes con alfa-talasemia es el portador silente en el 77.2% (34/44), seguido del rasgo alfatalasémico en 15.9% (7/44) y la enfermedad por Hb H en el 6.8% (3/44); estos últimos corresponden al porcentaje de pacientes que presentaron a la vez doble heterocigosis en el estudio molecular. No se reportaron casos de síndrome de hidrops fetalis en el periodo evaluado.
La coexistencia de alfa-talasemia y anemia ferropénica se determinó en el 9.0% (4/44) de los sujetos del estudio. Se demostró que el 84.1% (37/44) de los casos presentó una morfología de glóbulos rojos caracterizada por la hipocromía y microcitosis y el 15.9% (7/44) no mostraba alteraciones morfológicas.
El objetivo de este estudio es conocer las características clínicas y epidemiológicas de los pacientes con alfa-talasemia en la población pediátrica de Costa Rica mediante un estudio observacional descriptivo retrospectivo de revisión de expedientes. En Costa Rica se han realizado estudios de hemoglobinopatías anteriormente6; sin embargo, en la mayoría de ellos no se incluyen análisis moleculares para alfa-talasemia, o se deducen prevalencias de alfa-talasemia a partir de hallazgos de Hb H en electroforesis de hemoglobina, lo que limita su generalización en la población costarricense y su extrapolación a población infantil.
Un total de 60 pacientes satisface los criterios del estudio: diagnóstico de anemia (hemoglobina, menor de 12.0), morfología de glóbulos rojos caracterizada por hipocromía y microcitocis, (VCM menor de 80 fL y un HCM menor 27 pg), estudio electroforético con patrón normal para la hemoglobina y que incluye HbA2 normal (rango 2-3.3 %) o disminuida.
El 73.3% de los casos (44/60) tiene la condición de alfa-talasemia confirmada en el estudio molecular. La edad promedio de la población en estudio es de 4.9 años y en la distribución por sexo predominaron los hombres (55%) sobre las mujeres (45%).
En un estudio prospectivo efectuado en un hospital de Sri Lanka por S. Mettananda et al en 20207, en el que estudiaron a niños con anemia microcítica-hipocrómica para determinar talasemia y deficiencia de hierro, la distribución obtenida es similar a nuestros hallazgos: un 60.5% el sexo masculino y un 39.5% el femenino.
La observación elevada de anemia en el grupo de menores de 5 años correlaciona en este estudio con alfa-talasemia en el 71.6% de las muestras estudiadas, esto indica que es necesario que pacientes refractarios al tratamiento con hierro se estudien por sospecha de alfa-talasemia. En este grupo de edad se pudo constatar mayor afectación por anemia probablemente debida al crecimiento acelerado, con baja ingesta de hierro en la dieta e incluidos otros factores de riesgo como la prematuridad. 8
Todo esto fundamenta las estrategias realizadas por parte de las autoridades de salud en la implementación de programas de prevención y seguimiento de anemia en la edad preescolar (Caja Costarricense de Seguro Social. Manual de procedimientos para la atención integral del niño y la niña primer nivel de atención. San José: CCSS; 2016. [accesado 04-03-2020]. Disponible en https://es.scribd.com/document/350897693/Manual-Procedimientos-Atencion-del-Nino-a-I-Nivel-2016-final-2-pdf).
La provincia con mayor prevalencia de casos de alfa-talasemia confirmados por biología molecular es Limón, con prevalencia de 10.8; en segundo lugar, están las provincias de Guanacaste y Puntarenas con 8.5 casos por cada 100,000 habitantes en menores de 13 años. Lo anterior coincide con lo descrito en estudios nacionales9 que señalan que las hemoglobinopatías, incluyendo las alfa-talasemias, se observan con mayor frecuencia en poblaciones costeras, calculándose frecuencias que oscilan entre 6.1% y 18%. En Costa Rica, la población afrodescendiente representa un 1.05% de la población total, con mayor distribución en las provincias de Limón, Guanacaste y San José.10 Las costas son sitios de flujo migratorio constante con mayor posibilidad de mezclas étnicas, 7 lo que puede incidir en la frecuencia de distribución de las talasemias a nivel nacional, como lo reflejado en los datos presentados en las provincias de Guanacaste, Limón y Puntarenas.
Los fenotipos clínicos de la mayoría de las personas portadoras de alfa-talasemia son anemias de tipo leve-moderada asintomáticas; no se logra sospechar su condición hasta la realización de un hemograma de rutina.11 Por esta razón, en este estudio, al no generarse sospecha clínica de su condición, podría no haberse incluido niños con índices normales que pudieran tener genotipo de portador silente. Los niveles de hemoglobina encontrados en la población en estudio evidencian un promedio de hemoglobina de 10.4 g/dL con un rango entre 8.9 y 13.5 g/dL, lo que se correlaciona con la literatura.
Al agrupar los motivos de referencia de los pacientes participantes de este estudio, se obtuvo que la mayoría es referida por anemia hipocrómica microcítica en el 36.7% de los casos, y, en segundo lugar, por anemia con pobre respuesta al tratamiento con hierro. Tomando en consideración lo anterior, es importante que se establezca un diagnóstico diferencial utilizando la biología molecular para hallar las alfa-talasemias en pacientes que se presenten con una anemia de características hipocrómicas microcíticas y refractarios al tratamiento con hierro oral supervisado y a dosis óptimas. Lo anterior, guiado bajo una anamnesis adecuada (no olvidando los antecedentes de herencia familiar) y un examen físico completo; lo que podría dar oportunidad a la derivación temprana a un especialista y por ende al diagnóstico temprano de alfa-talasemia.
El patrón de herencia mendeliana es un aspecto trascendental en la presentación y distribución de las enfermedades genéticas, siendo que las alfa-talasemias consisten en enfermedades genéticas de herencia autosómica recesiva12 en las que son necesarias dos copias de la mutación del gen para producir la enfermedad.
La existencia de antecedente de hemoglobinopatía en familiares se reporta en el 36.3% de los niños del estudio confirmados con alfa-talasemia; en contraste con reportes de un 4.8% de antecedente heredofamiliar en la historia clínica de pacientes pediátricos, según el estudio de S. Mettananda et al en 2020 7 realizado en Sri Lanka, cuyo objetivo fue analizar estrategias de prevención de talasemia en países de bajos a medianos ingresos. Es importante realizar un mayor reporte de antecedentes familiares en los expedientes clínicos, pues en muchos casos el médico podría no haberlo anotado en el expediente y generar con ello un bajo porcentaje de antecedentes heredo-familiares en este estudio. La concientización del médico ante casos de anemia refractaria ayudará a la búsqueda más dirigida en familias y facilitará así la detección de casos de alfa-talasemia.
La presentación clínica más evidente y de severidad intermedia en pacientes con alfatalasemia va a estar condicionada a la afectación de 2 alelos del mismo cromosoma (a0). En este estudio, el tipo más frecuentemente encontrado según el número de alelos afectados es a+ (pérdida de un solo alelo), por lo que se constata que el 77.2% de la población estudiada no tiene una presentación clínica evidente, más que los hallazgos del hemograma (Cuadro 1) o la refractariedad al tratamiento con hierro. El fenotipo de alfatalasemia más numeroso en la muestra estudiada es el portador silente, con el 77.2% de los casos, cifra que coincide con otros estudios en América en los que el hallazgo genotípico más frecuente es el portador silente por deleción -3.7 Kb 13 y en zonas de alta frecuencia como en el Oriente, en donde también es una presentación clínica que lidera dentro de la población general.14 El portador silente es asintomático y los hallazgos en el hemograma, de encontrarse, suelen ser sutiles, lo que puede dirigir a un diagnóstico erróneo con la anemia por carencia de hierro y a que se ofrezca un abordaje incorrecto con una terapéutica con hierro oral innecesaria y que esto signifique un retraso en la derivación temprana a un especialista.
Uno de los diagnósticos diferenciales de las alfa-talasemias, por su forma de presentación en los parámetros hematológicos encontrados, es la anemia por deficiencia de hierro (Caja Costarricense de Seguro Social. Abordaje de la Anemia por Deficiencia de Hierro en niños y niñas de 6 a 24 meses de edad de Costa Rica en el año 2014. Víquez M. [accesado 16-05-2021]. Disponible en: https://www.binasss.sa.cr/serviciosdesalud/ anemiahierro2014.pdf). En este estudio se evidencia la coexistencia de alfa-talasemia y anemia ferropénica en un 9% de los pacientes (4/44), datos similares a otros estudios 7 que encontraron que un 6.7% de su población presentaba coexistencia entre estas dos entidades. Los pacientes con alfa-talasemia no están exentos de tener anemia por deficiencia de hierro y esta coexistencia puede hacer más evidente el descenso en los parámetros hematológicos y acompañarse de poca respuesta a la terapia con hierro puesto que dicha coexistencia entre alfa-talasemia y anemia por deficiencia de hierro es un hallazgo común.
Son numerosos los estudios que exponen la frecuencia de la presentación de la alfa-talasemia en el hemograma con eritrocitosis acompañada de hipocromía y microcitosis, como características indicativas de sospecha de talasemia.13,14,15 Para la población analizada, se obtuvo una media de recuento de glóbulos rojos en 5.22 (x106/μL), VCM 65.3 fL y HCM en 21.4 pg, lo que se correlaciona con lo expuesto en la literatura y en estudios previos16 que sugieren la presencia de perfiles hematológicos definidos con recuento de glóbulos rojos mayor o igual a 4.5 106/mm3en las alfa-talasemias, así como VCM menor a 80 fL y HCM menor a 27 pg. Cabe destacar que el 84.1% de los casos confirmados de alfa-talasemia presentó morfología de glóbulos rojos hipocrómica microcítica en el hemograma inicial, siendo todos los parámetros anteriores elementos importantes para consideración por parte de los médicos en el proceso de evaluación diagnóstica de estos pacientes.17, 18
La confirmación de los casos mediante el estudio molecular que consistió en la identificación las 21 mutaciones/deleciones más frecuentes del gen de a globina por un método de amplificación por PCR e hibridación inversa4 confirmó que el 73.3% de los niños (44/60) presentaba afectación de alguno de los genes de la molécula de alfaglobina, mientras que en los 16 pacientes restantes no se logró determinar su causa. Por la metodología utilizada, un resultado negativo no excluye la enfermedad, pues podrían no estarse analizando regiones del genoma que no están contempladas en el kit utilizado. En el período de estudio, solo se contaba con la metodología mencionada, por lo que se debe considerar que en la literatura se han descrito más de 100 genotipos distintos a nivel global de alfa-talasemia que no fueron analizados para estos pacientes.1
Los defectos genéticos de las alfa-talasemias son con mayor frecuencia resultado de defectos delecionales de uno o más genes del cromosoma. Ocasionalmente, se deben a defectos mutacionales, también conocidos como variantes no delecionales. 1 En nuestro estudio, se evidenció que de los 44 casos de a talasemia confirmados mediante estudio molecular, el 95.5% era representado por deleciones de genes a y un 4.5% por variante no delecional. Estos resultados comparten similitudes con los obtenidos en otros estudios internacionales, como en España19, en los que se obtuvieron cifras de hasta un 92,3% de alfa-talasemia delecional frente a 7.7% de variante no delecional.13,14 En Oriente Medio se reportan mayores porcentajes de presentación de casos delecionales, pero con una ligera discrepancia en las variantes no delecionales. Las variantes no delecionales en otras zonas del mundo, como Irán14 y Turquía,13 se reportan en el 32.7% y 30.6% de casos respectivamente, esta diferencia puede deberse a que son zonas geográficas con especial concentración de alfa-talasemia distribuidas en el Mediterráneo y regiones del Sudeste Asiático, lugares en que se encuentran portadores hasta en un 40% de la población, 13 una mayor prevalencia de portadores de alfa-talasemia en comparación a nuestro país.
La deleción -3.7 Kb fue el genotipo más frecuente encontrado en este estudio; agrupó un 77.2% de los casos. Este es el defecto genético más comúnmente evidenciado en los estudios tanto de Oriente Medio13,14 como en los realizados en el continente americano.16 Se ha reportado una amplia distribución a nivel mundial, siendo la variante delecional la más frecuente en muchos grupos poblacionales.17 La segunda variante delecional más frecuente es la SEA, presente en niños de ascendencia asiática. Estos datos son de suma importancia ya que, hasta la realización de este estudio, no se contaba con la descripción de las deleciones más frecuentes en Costa Rica para alfatalasemia. Costa Rica no es considerada una zona endémica de alfa-talasemia, pero su conformación poblacional en los últimos años ha cambiado debido a la alta migración, con reportes de hasta un 9.0% de personas migrantes residentes en el país, de las cuales cerca del 0.2% es representado por población de origen oriental.10 Es así como la frecuencia de presentación de otros tipos delecionales distintos a la variante delecional -3.7 Kb es cada vez más común de encontrar en la población costarricense.
Por otro lado, las variantes mutacionales en las alfa-talasemias correspondieron al 4.5% de los casos, con evidencia de 2 casos de doble heterocigotas para hemoglobina Constant Spring, siendo esta la mutación más frecuente en países asiáticos. Cabe mencionar que estos pacientes con la condición de doble heterocigosis corresponden a casos de enfermedad por Hb H.
Para el presente estudio no se encontraron otras deleciones como la del tipo --MED (frecuente en individuos de ascendencia mediterránea) ni a2polyA1, las cuales alcanzan reportes de hasta el 17.9% y 6.3% respectivamente en países como Turquía.13
Según lo descrito en la literatura cuando se detectan variantes de hemoglobina en las alfatalasemias, por lo general se debe a genotipos homocigotos o heterocigotos para mutaciones y deleciones del gen de alfa-globina.10 En este estudio se reportó una distribución muy similar en los tipos de cigocidad: 47.7% para heterocigotos y 47.7% para homocigotos (pacientes comprendidos entre enero del 2018 y enero del 2019). Este es un hallazgo de la epidemiología de Costa Rica que hasta la fecha se desconocía; sin embargo, sería necesario ampliar el estudio a mayor número de pacientes.
Por todo lo expuesto, se puede concluir que el hemograma es una útil herramienta de evaluación inicial que, tomando en cuenta los hallazgos demostrados en este estudio (eritrocitosis con microcitosis e hipocromía en un paciente refractario al hierro), debe sentar la sospecha inicial de alfatalasemia y que, junto con una electroforesis de hemoglobina reportada como normal, permitiría realizar una derivación temprana para el oportuno abordaje de esta enfermedad.
Finalmente, el movimiento migratorio actual de turistas y residentes hace que se presenten con mayor frecuencia entrecruzamientos étnicos resultantes en genotipos talasémicos poco frecuentes en nuestra población y evidenciados por los dos casos dobles heterocigotos por SEA y Hb Constan Spring ya mencionados.
Con la información obtenida en este año de estudio, en el que se describen las variantes más frecuentes de las alfa-talasemias en población pediátrica de Costa Rica, se puede brindar una perspectiva actualizada en torno a la enfermedad presente en los niños costarricenses, información con la que no se contaba previamente. Sin embargo, el estudio contó con la limitante de no poder calcular la prevalencia general en la población costarricense, pues no se incluyó a pacientes de otros centros de salud ni de todas las edades ni de muchos años pues para ello se habría requerido de un estudio aleatorizado que contemplara población general. Aunque en el presente estudio solo hubo 3 casos de alfa-talasemia con pérdida de tres de los cuatro genes alfa (enfermedad por Hb H), estos son los pacientes con mayor riesgo de heredar formas severas como hidrops fetalis a su descendencia y a los cuales es de suma trascendencia brindarles un diagnóstico precoz para su evaluación y manejo especializado.
El diagnóstico oportuno de estos sujetos asegura un seguimiento a largo plazo y consejería genética para determinar el riesgo reproductivo de las parejas2 con el fin de reducir las complicaciones tanto maternas como fetales asociadas a la enfermedad y la aparición de nuevos casos.
Este estudio es el primer reporte epidemiológico a nivel nacional de pacientes pediátricos con alfa-talasemia y aporta datos estadísticos antes desconocidos y permite al médico una guía para la sospecha de esta patología.
Los índices hematimétricos del hemograma y la electroforesis de hemoglobina, junto con la sospecha clínica, pueden brindar datos sugestivos de alfa-talasemia, pero para su confirmación es indispensable el estudio molecular. Dentro de los métodos moleculares que se realizan en el país están la reacción en cadena de polimerasa (PCR) y la amplificación de sondas dependiente de ligandos múltiples (MLPA, por sus siglas en inglés), que son los métodos más utilizados para detección de deleciones; la secuenciación de nueva generación (NGS, por sus siglas en inglés) se utiliza para mutaciones puntuales o deleciones cortas.3,4
Cabe mencionar que, actualmente, en Costa Rica, el análisis molecular por alfa-talasemia no forma parte del Programa Nacional de Tamizaje Neonatal Nacional; no obstante, los casos aislados de detección Hb H son referidos al Laboratorio de Investigación del Hospital de Niños para estudio del gen de alfa-globina para su caracterización y al servicio de Hematología para su seguimiento.

