









Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkActa Médica Costarricense
versão On-line ISSN 0001-6002versão impressa ISSN 0001-6012
Acta méd. costarric vol.56 no.3 San José Jul./Set. 2014
Caso Clínico
Diabetes mellitus neonatal en Costa Rica
Neonatal Diabetes Mellitus in Costa Rica
Francis Ruiz-Salazar,1* Erick Richmond-Padilla,2* Roberto Bogarín-Solano,2 Fred Cavallo-Aita2 y Orlando Jaramillo-Lines2
Resumen
La diabetes mellitus neonatal es un raro desorden metabólico usualmente desarrollado en las primeras 6 semanas de vida, secundario a un grupo de mutaciones y defectos del desarrollo pancreático que puede desembocar en una catástrofe clínica si no se identifica tempranamente; se divide en una variante transitoria y una permanente, siendo la primera la más frecuente, con cerca de un 60% de los casos. El manejo inicial de ambas variantes es la insulinoterapia intensiva, que en la variante transitoria puede suspenderse usualmente en los primeros meses de vida. El síndrome de disregulación inmunológica, poliendocrinopatía y enteropatía ligada a X (IPEX por sus siglas en inglés), es una causa extremadamente rara de la variante permanente, casi siempre mortal, caracterizada por un inmunocompromiso severo, enteropatía, diabetes y dermatitis. En el estudio se describen 4 casos de diabetes mellitus neonatal diagnosticados en el Hospital Nacional de Niños de San José, Costa Rica: 2 correspondientes a una diabetes mellitus neonatal transitoria,
Descriptores: diabetes neonatal Costa Rica, diabetes neonatal transitoria, diabetes neonatal permanente, síndrome de IPEX
Abstract
Neonatal diabetes mellitus s a rare metabolic disease that usually develops in the first 6 weeks of life. It is caused by a group of mutations and birth defects in the organogenesis of the pancreas that could be catastrophic if not identified early. It is divided in a transient and a permanent variant, the first one is more frequent as it is related to 60% of cases. The initial treatment in both variants is intensive insulin therapy, but in the transient variant it may be suspended in the first months of life. IPEX syndrome is an extremely rare cause of permanent neonatal diabetes, usually mortal, characterized by severe immunological compromise, enteropathy, diabetes and dermatitis. In this study we describe four cases of NDM diagnosed at the National Children’s Hospital in
Keywords: Diabetes neonatal in Costa Rica, transitory neonatal diabetes, permanent neonatal diabetes, IPEX syndrome.
La diabetes mellitus neonatal (DMN) es un desorden metabólico infrecuente que se presenta en las primeras semanas de vida, relacionado con niveles bajos de insulina e hiperglicemia. Es causado por una variedad de mutaciones y defectos del desarrollo del páncreas que modifican el curso clínico, según su presencia, en una variante transitoria y una permanente,1 siendo la primera la más frecuente, con cerca de un 60% de los casos. El manejo inicial de ambas variantes es la insulinoterapia intensiva, que en la variante transitoria puede suspenderse usualmente en los primeros meses de vida. El síndrome de desregulación inmunológica, poliendocrinopatía y enteropatía ligada a X (IPEX por sus siglas en inglés), es una causa extremadamente rara de la variante permanente, casi siempre mortal, caracterizada por un inmunocompromiso severo, enteropatía, diabetes y dermatitis. En el estudio se describen 4 casos de DMN diagnosticados en el Hospital Nacional de Niños de San José, Costa Rica: 2 correspondientes a una DMN transitoria,
Reporte de casos
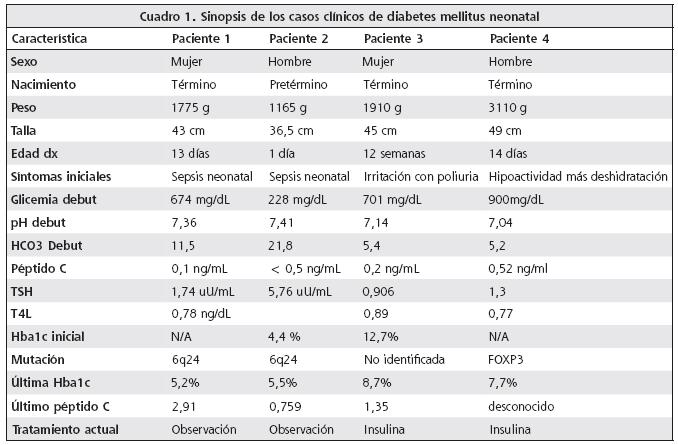
Paciente 1.
Femenina que nace a las 38 semanas de edad gestacional, sin complicaciones periparto, que debutó a los 13 días con un cuadro de sepsis neonatal caracterizado por vómitos y fiebre. Se describe inicialmente con mal estado general asociado a deshidratación moderada; análisis de laboratorio reportó una glicemia en 674mg/ dL, además de glucosuria sin cetonemia y una acidosis metabólica moderada sin acidemia. Comienza esquema intensificado con insulina lispro 8 dosis al día, con lo que se controla de manera adecuada. Se mantuvo internada por 3 semanas y se egresa con dosis de insulina lispro menores a 1U por día. En colaboración con el laboratorio genético regional de Exeter, Reino Unido, se documenta la presencia de la mutación del locus 6q24, y se confirma el diagnóstico de diabetes neonatal transitoria. Durante el seguimiento en la consulta externa se logra suspender la insulina ultrarrápida a partir de los 4 meses de edad.
Paciente 2.
Masculino que nace a las 34 semanas por cesárea, debido a un retardo del crecimiento intrauterino (RCIU) asociado a hipertensión materna inducida por el embarazo; nace sin complicaciones, pero desarrolla sepsis neonatal en los primeros 5 días. En control bioquímico se le documentan glicemias entre 200 y 300 mg/dL en forma repetida, por lo que se sospecha el diagnóstico de diabetes neonatal e inicia esquema intensificado con lispro 8 dosis diarias. Su internamiento duró 7 semanas debido a cuadro de sepsis nosocomial por E. faecalis. Durante el seguimiento en consulta externa se disminuyen gradualmente requerimientos insulínicos hasta su suspensión completa a partir del año del diagnóstico. Se logra de igual forma, la confirmación mediante análisis genético, de la mutación 6q24. Como comorbilidad asociada se diagnostica epilepsia parcial criptogénica.
Paciente 3.
Femenina que nace a las 40 semanas, pequeña para edad gestacional, con puntaje APGAR 6-8; no se documentan complicaciones. Consulta a los 3 meses de edad al Servicio de Emergencias, por un cuadro agudo de 3 días de evolución de irritabilidad, poliuria y resequedad oral. Se describe inicialmente con deshidratación severa, taquicardia y franca disnea; laboratorios iniciales señalan una glicemia en 701 mg/dL asociada a una acidosis metabólica severa con acidemia, cetonemia y cetonuria importantes. Se le realiza una hemoglobina glicosilada que se reporta en el 12,7%, además de una insulinemia indetectable; se le inicia esquema insulínico con lispro 8 dosis diarias. Se mantuvo hospitalizada por 2 semanas. No se identificaron anticuerpos antiGAD ni antiinsulina. Tomografía axial computarizada realizada a nivel abdominal reportó una hipoplasia pancreática. Se le efectúa estudio de mutaciones causales de diabetes neonatal permanente, en específico kir 6,2, SUR1 (ABCC8) e INS Exones 1-3, sin encontrar compromiso, por lo que también se practica estudio sobre región 6q, en búsqueda de mutaciones asociadas a diabetes neonatal transitoria, sin encontrar ninguna de las mutaciones asociadas. Ha tenido una evolución satisfactoria, pero no se ha logrado disminuir los requerimientos insulínicos.
Paciente 4.
Masculino que nace a las 38,5 semanas, considerado adecuado para la edad gestacional, parto sin complicaciones, embarazo de bajo riesgo. A los 14 días de edad presenta un cuadro de hipoactividad, pobre succión y deshidratación. En el Servicio de Emergencias le documentan hiperglicemia y cetacidosis severa. Se controla y maneja con esquema intensivo de lispro. Tomografía de abdomen reporta un páncreas de tamaño y forma normales. AntiGAD y antiinsulina negativos. Se egresa con el diagnóstico de diabetes mellitus neonatal y manejo con lispro.
Posteriormente, el paciente es valorado en múltiples ocasiones por cuadros diarreicos asociados a pérdida de peso. Debido a la severidad y cronicidad de este caso, se inician estudios por enteropatía autoinmune; se diagnostica hipogammaglobulinemia y comienza gammaglobulina y esteroides y azatioprina; se determina a los 9 meses la presencia de hipotiroidismo e inicia levotiroxina. A los 15 meses el paciente es ingresado por un cuadro de hepatitis que se concluyó ser de origen autoinmune. Por los trastornos inmunológicos, diabetes neonatal, hipotiroidismo y enteropatía crónica, se plantea la posibilidad de un síndrome de IPEX, por lo que se realizan estudios moleculares en el laboratorio de Genética Humana del Ospedali Galliera di Genova, Italia. Se concluye que el paciente es homocigota para la mutación IVS1+2T>G del gen FOXP3, con lo que se confirma el diagnóstico de síndrome de IPEX.
A los 18 meses se le realiza trasplante de médula ósea alogénico; el donador fue su hermano. El paciente ha tenido una evolución excelente, su desarrollo global es normal, posee una talla y peso adecuados y mantiene su control glicémico en las metas esperadas para su edad, con el uso de un esquema insulínico intensificado.
En la presentación de los pacientes (Cuadro 1) se describe 2 casos con diabetes mellitus neonatal transitoria, portadores de la mutación 6q24. Ambos refuerzan la evidencia de la bibliografía sobre esta enfermedad, ya que su curso clínico es clásico; los 2 pacientes presentaron RCIU, debutaron en los primeros días de vida sin desarrollar cetoacidosis diabética, su inmunología fue negativa por diabetes mellitus tipo 1, y se logró suspender en ellos el uso de insulina antes del año de edad. Los 2 últimos casos corresponden a pacientes con diabetes mellitus neonatal permanente, 1 asociado a hipoplasia pancreática y el otro secundario a un raro síndrome conocido como IPEX. Ambos pacientes debutaron con cuadro de cetoacidosis diabética, no hubo evidencia de RCIU y ninguno ha logrado disminuir sus requerimientos de insulina. En el paciente 3 no se logró identificar ninguna de las mutaciones descritas en los casos transitorios ni permanentes, situación identificada en los pacientes con anomalías en el desarrollo pancreático. El paciente 4 es un excelente ejemplo del síndrome de IPEX: es un hombre, debutó con DMN, desarrolló cuadros diarreicos severos difíciles de controlar, se diagnosticó tempranamente con hipotiroidismo, presentó varios episodios de sepsis y su curso clínico mejoró significativamente con el trasplante alogénico de médula ósea, tal y como ha sido descrito en otros reportes.
Revisión bibliográfica
La DMN es un raro desorden no inmunológico que conlleva un potencial desastre metabólico para el neonato; se caracteriza por hiperglicemia asociada a niveles bajos de insulina. Su incidencia oscila entre 1:300
La DMN se divide en aquellas que poseen un curso transitorio y en las que, por el contrario, son permanentes. La DMN transitoria se define como la que inicia en las primeras 6 semanas de vida, pero que se recupera antes de los 18 meses.4 Es atribuible en la mayoría de los casos a defectos en el marcaje genético en el cromosoma 6, mientras que las permanentes son el resultado de anormalidades en el desarrollo pancreático, como una hipoplasia, o agenesia, o defectos genéticos en los canales de potasio ATP dependientes.5
Patogénesis de la diabetes mellitus neonatal
Diabetes mellitus neonatal transitoria (DMNT): la DMN transitoria es usualmente de presentación esporádica, aunque la trasmisión paterna ha sido documentada en cerca de una tercera parte de los casos, en ausencia, inclusive, de diabetes mellitus en los padres. Isodisomía paterna, duplicación incompleta del brazo largo del cromosoma 6q24, o una anomalía en la desmetilación, son los defectos encontrados.6 En una cohorte de pacientes portadores de DMN transitoria, se documentó que de los 30 casos reportados, 23 eran esporádicos y 7 familiares; del total se identificó que el 50% tenía una isodisomía paterna; el 20%, una duplicación y el 8%, un defecto de metilación.7 Para que estos defectos generen DMN deben ser transmitidos por el padre, lo cual indica que el mecanismo de enfermedad es el marcaje genético. Estos defectos relacionados con el cromosoma 6q24 son la principal causa de la DMN transitoria, siendo la mutación del ZAC/PLAG1, la más frecuente de todas. PLAG1 es el gen de un receptor relacionado con la célula beta, cuyo ligando es un potente secretagogo de insulina. ZAC, por su parte, es un regulador del ciclo celular y la apoptosis, alterando el número absoluto y eficiencia de la célula beta del páncreas in útero.8
El marcaje genético o “imprinting”, como se denomina en inglés, es una expresión diferencial de un material genético dependiente de si fue heredado por el padre o la madre; un marcaje materno indica que el alelo heredado por la madre se inactiva, expresando por tanto solo el material del alelo paterno. En el caso de la DMN transitoria, la trasmisión de la isodisomia o la duplicación del brazo largo del cromosoma 6 por parte del padre, generará la enfermedad, ya que al existir un marcaje materno, a pesar de que el alelo de la madre sea normal, será inactivado.
Desde el punto de vista clínico, la DMNT es responsable de entre el 50 y el 60% de los casos de DMN.9 Suelen presentarse con RCIU asociado a la deficiencia de insulina como factor de crecimiento, principalmente en el tercer trimestre. Se caracterizan por debutar en los primeros días de vida; a pesar de no tener la inmunología clásica de un DM tipo 1, pueden desarrollar deshidratación severa y cetoacidosis, aunque en una proporción significativamente menor que los pacientes con DMNP;10 posterior al debut, los requerimientos insulínicos empiezan a reducirse en forma progresiva, hasta suspender esta alrededor del año de edad.11 El curso clínico posterior requiere vigilancia, debido a que se ha observado que la gran mayoría, cerca de dos terceras partes de los pacientes, hacen recurrencia de la diabetes en los años posteriores, usualmente alrededor de la adolescencia, por lo que se ha planteado a la resistencia insulínica de la pubertad como un factor detonante, aunque no ha sido demostrada. Se desconoce de igual forma el motivo de la remisión inicial, pero se ha sugerido la expansión de la célula beta en el periodo neonatal, como evento responsable.12,13 El concepto actual es que la DMNT es realmente una variante permanente con un curso clínico más larvado.
Diabetes mellitus neonatal permanente: alrededor del 30 al 40% de los casos de DMN son permanentes; la gran mayoría de estos pacientes presentará mutaciones del canal de potasio ATP dependiente, mientras que los defectos del desarrollo pancreático ocurren rara vez. Mutaciones en el gen de la glucokinasa también han sido reportadas como causa de DMN permanente.4
El canal de potasio ATP dependiente es un compuesto octamérico formado por una pequeña subunidad Kir6.2 y una subunidad SUR1; su acción regula la liberación de insulina; al cerrarse permite la despolarización de la célula beta y al abrirse, la hiperpolarización. Mutaciones activantes del receptor favorecen la apertura de estos canales, con lo cual se evita la despolarización y, por lo tanto, la liberación de insulina, con lo que se induce la DMN.14
La mutación activante del gen KCNJ11, encargado de codificar la subunidad Kir6.2, es la causa más común de DMN permanente. Otra de las causas importantes es la mutación activante de la subunidad SUR1 codificada por el gen ABCC8; esta mutación tiene la particularidad de producir tanto DMN transitoria como permanente. Por ejemplo, un estudio en pacientes con DMN encontró que de los 73 casos, 9 tenían la mutación ABCC8, y de esos solo 2 fueron permanentes.15
Los casos reportados de agenesia pancreática en la bibliografía son escasos. Para 2008, Chen et al5 contabilizaron 23 casos. De estos, solamente en 6 se logró demostrar un defecto genético a nivel de Pdx1, un factor de transcripción relacionado con el desarrollo pancreático. En los demás casos no se logró identificar una mutación causal. Se ha descrito en modelos animales, factores de transcripción adicionales, como: Sox17, Sox9, Hlxb9, Ptf1a, Hnf6, RfX6, EIF2AK3, GLIS3 y FOXP3, entre otros;16 el último se relaciona con el raro síndrome de IPEX.
Clínicamente, establecer la diferencia entre una DMN permanente y una transitoria, al momento del debut, no es fácil, aunque la menor presencia de RCIU y la mayor incidencia de cetoacidosis en las formas permanentes, ayudan a orientar el diagnóstico. Al igual que en los casos transitorios, la inmunología y los HLA no se relacionan con la DM tipo 1.
Tratamiento
El tratamiento inicial de ambas variantes de la DMN es la insulina. A pesar de ser un grupo de pacientes muy difíciles de manejar, no existe suficiente evidencia para establecer un esquema como el estándar de cuidado. Existen grupos que recomiendan un esquema enfocado en insulinas basales, pero la variabilidad de las insulinas humanas aumenta en forma peligrosa el riesgo de hipoglicemias, y los análogos no están aprobados en este contexto. La administración de múltiples dosis de insulinas rápidas y ultrarrápidas ha sido reportada en diferentes casos como efectiva, especialmente en el control inicial de la enfermedad. Un grupo francés, por su parte, ha publicado resultados positivos con el inicio temprano de bombas de infusión tras la estabilización primera con el uso de insulina endovenosa.11
En el caso de la DMNP, en especial la producida por la mutación del gen KCNJ11, el uso de las sulfonilureas ha demostrado ser eficaz al permitir controlar adecuadamente la enfermedad y, a su vez, posibilitar la suspensión de la insulina. La sulfonilurea, al cerrar el canal de potasio ATP dependiente, permite liberar la insulina pancreática y vencer de esta manera la mutación activante del receptor. Un estudio francés en 49 casos de DMNP debido a la mutación KCNJ11, demostró que el traslape a glibenclamida permitió suspender la insulina en el 90% de los casos, y que el control glicémico mejoró de una Hba1c del 8,4% al 6,8%, en cerca de 12 semanas.17
Síndrome de IPEX
El síndrome de IPEX es el acrónimo de disregulación inmunológica, poliendocrinopatía y enteropatía ligada a X; es un raro síndrome, frecuentemente fatal, que se presenta con la triada de enteropatía, endocrinopatía autoinmune y dermatitis. El compromiso inmunológico es el evento determinante en esta enfermedad.17
FOXP3 es el gen mutado en el síndrome de IPEX. Este se encarga de la producción de un factor de transcripción fundamental para los linfocitos T reguladores, cuya función es la de ser un potente inmunosupresor y permitir la autotolerancia. También se cree que regula la diferenciación de estos linfocitos.18 Se localiza en el cromosoma Xp11.23 y se han descrito al menos 20 mutaciones, la mayoría de ellas familiares, aunque casos esporádicos han sido reportados.19,20 No existe una correlación adecuada entre el genotipo y el fenotipo.
La prevalencia del IPEX se desconoce, debido a que es un padecimiento sumamente raro y con frecuencia mortal, lo cual lo hace susceptible a errores de diagnóstico, o a no ser diagnosticado del todo. Un reporte austriaco sobre diabetes neonatal, identificó un solo caso de IPEX para una incidencia de 1 por cada 1 609 490.21 Al ser de herencia ligada al X, los pacientes afectados son hombres, mientras que las mujeres son portadoras asintomáticas; sin embargo, se ha descrito en una familia la presencia de IPEX, sin identificar la mutación en FOXP3 y, además, la presencia de la enfermedad en una mujer indica una heterogeneidad genética mayor que la inicialmente descrita.22
EL IPEX tiende a presentarse en el periodo perinatal como una diabetes mellitus o una diarrea secretoria crónica; la presencia de una dermatitis eczematosa completa la triada clásicamente descrita. La mayoría de estos pacientes tiene falla para progresar, procesos infecciosos a repetición con respuestas exageradas, citopenias, alergias a múltiples alimentos y tiroiditis. El curso clínico habitual es el desarrollo de crisis provocadas por infecciones, alérgenos y otros detonantes inmunológicos, principalmente en los pacientes sin trasplante de médula ósea.
Establecer el diagnóstico de IPEX depende de una alta sospecha clínica. Se debe plantear especialmente en aquellos pacientes masculinos que debutan con diabetes neonatal o desarrollan una diarrea crónica de difícil manejo; la presencia de dermatitis, hipotiroidismo e infecciones, aumenta aún más la sospecha, pero el diagnóstico definitivo se confirma con la biología molecular positiva por el FOXP3. Hallazgos adicionales, como autoanticuerpos contra la tiroides, páncreas y eritrocitos, son frecuentes.23
El tratamiento del IPEX se puede dividir en 2: control de crisis y tratamiento crónico con inmunosupresores y dieta. El manejo de la diabetes mellitus y sus descompensaciones no dista en forma significativa del abordaje clásico. El control de las crisis requiere aislamiento y optimizar la inmunosupresión. En ocasiones, dichas descompensaciones pueden simular infecciones, por lo que la decisión de tratamiento siempre debe individualizarse. Dosis elevadas de glucocorticoides entre 1-2mg/kg, son frecuentemente necesarias. Las crisis de enteropatía, manifestadas por diarreas de difícil control, ameritan a veces hospitalizaciones y nutrición parenteral total como medida salvadora. El tratamiento crónico se basa en el uso de glucocorticoides y otros inmunosupresores, como el sirolimus y los inhibidores de calcineurina, como el tacrólimus o ciclosporina. No existe estudios que hayan demostrado la superioridad de un esquema sobre otro; el esquema inicial con esteroides lo que busca es estabilizar la enfermedad para posteriormente traslaparlos hacia agentes como el sirolimus. El trasplante alogénico de médula ósea es el único procedimiento curativo conocido; la experiencia es limitada, pero se considera que cuanto a menor edad se pueda realizar el trasplante, menor riesgo existirá de desarrollar las complicaciones en forma permanente; de hecho, se ha determinado que muchas de las características del IPEX remiten luego al trasplante, con la excepción de la diabetes y el hipotiroidismo.24
En cuanto al control nutricional, el concepto clave es la identificación de alimentos que le produzcan brotes alérgicos al paciente, ya que son detonantes importantes de las crisis; todo paciente con IPEX debe, por lo tanto, tener un control con un especialista en esta área. Otro de los cuidados especiales consiste en evitar la aplicación de vacunas para evitar una respuesta inmunológica exagerada, a menos que sea estrictamente necesario. Se ha postulado aumentar en forma transitoria la inmunosupresión, pero eso disminuye a su vez la eficacia de la vacuna.
A pesar de que el caso presentado en nuestro servicio ha logrado una excelente evolución, la realidad es que el pronóstico de estos pacientes es malo; por ejemplo, una serie de pacientes documentó que de los 51 casos identificados, 23 ya habían fallecido. Muchos de estos pacientes mueren en los primeros años de vida, principalmente por cuadros de malabsorción, falla para progresar e infecciones.25
La DMN es una entidad clínica infrecuente, pero potencialmente mortal, que requiere de un diagnóstico oportuno. A pesar de su clasificación como transitoria y permanente, la evidencia parece demostrar que eventualmente algunos de los pacientes catalogados como transitorios terminan desarrollando una diabetes permanente, por lo que el seguimiento es fundamental. Aunque su manejo inicial es difícil, no existe un consenso en cuanto a cuál debería ser el estándar de cuidado en estos pacientes. Finalmente, el síndrome de IPEX es una causa extremadamente rara de DMN permanente, que precisa un manejo interdisciplinario y agresivo, por la alta mortalidad asociada; el único abordaje curativo conocido es el trasplante alogénico de médula ósea.
Conflicto de interés: ninguno.
Referencias
1. Polak, M. Neonatal diabetes mellitus: a disease linked to multiple mechanisms. Orphanet J Rare Dis 2007; 2:1750-1172
2. Shield J, Gardner RJ,
3. Kalhan SC, Devaskar Su. Neonatal-Perinatal Medicine: Diseases of the Fetus and Infant. 2011. Vol 2, 1497-1508. [ Links ]
4. Temple I. Transient neonatal diabetes, a disorder of imprinting. J Med Genet 2002;39:872–875. [ Links ]
5. Chen R, Hussain K, Al-Ali M,
6. Cave H, Polak M, Drunat S, Denamur E, Czernichow P: Refinementof the 6q chromosomal region implicated in transient neonatal diabetes. Diabetes 2000, 49:108-113. [ Links ]
7.
8. Kamiya M, Judson H,
9. Von Muhlendahl KE, Herkenhoff H: Long-term course of neonatal diabetes. N Engl J Med 1995, 333:704-708. [ Links ]
10. Flechtner I. Neonatal hyperglycaemia and abnormal development of the páncreas. 2008. Best Pract Res Clin Endocrinol Metab. 22:1;17–40. [ Links ]
11. Shield JP, Baum JD: Transient neonatal diabetes and later onsetdiabetes: a case of inherited insulin resistance. Arch Dis Child 1995, 72:56-57. [ Links ]
12. Greeley S, Tucker S, Naylor R, Bell G, Philipson L. Neonatal diabetes mellitus: A model for personalized medicine. Trends Endocrinol Metab. 2010. 21:464– 472. [ Links ]
13. Metz C, Cave H, Bertrand AM, Deffert C, Gueguen-Giroux B, Czernichow P, Polak M: Neonatal diabetes mellitus: chromosomal analysis in transient and permanent cases. J Pediatr 2002,141:483-489. [ Links ]
14. Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004; 350:1838. [ Links ]
15. Babenko AP, Polak M, Cavé H. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 2006; 355:456. [ Links ]
16. Scharfmann R, Polak M. Transcribing neonatal diabetes mellitus. N Engl J Med 2010; 362:1538. [ Links ]
17. Powell B. Buist N. Stenzel P. An X linked síndrome of diarrea, polyendocrinopathy and fatal infection in infancy. The Journal of Pediatrics, 1982; 100:731-737.
18. Chatila TA. Role of regulatory T cells in human diseases. J Allergy Clin Immunol 2005; 116:949.
19. Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol 2007; 120:744.
20. Hans J. Van der Vliet. Nieuwenhuis E. IPEX as a Result of Mutations in FOXP3. Clin Dev Immunol. 2007;2007:89017:1-5.
21. Wiedemann B, Schober E, Waldhoer T, Koehle J, Flanagan SE, Mackay DJG et al. Incidence of neonatal diabetes in Austria-calculation based on the Austrian Diabetes Register. Pediatr Diabetes 2010; 11:18.
22. Wildin R. Smyk-Pearson S. Filipovich A. Clinical and molecular features of the Immune dysregulation, polyendocrinopathy, enteropathy, X-linked síndrome. J Med Genet., 2002. 39:8 pp 537-545.
23. Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D et al. Treatment of the Immune dysregulation, polyendocrinopathy, enteropathy, X-linked síndrome by allogenic bone marrow transplantation. 2001. N Engl J Med 344:23, 1758-1762.
24. Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning, 2007. Blood. 109:383-385.
25. Ochs HD, Torgerson TR. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked inheritance: model for autoaggression. Adv Exp Med Biol. 2007; 601:27-35.
Trabajo realizado en el Servicio de Endocrinología Pediátrica del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”
Afiliación de los autores: 1Servicio de Endocrinología Hospital México y
2Servicio de Endocrinología Pediátrica, Hospital Nacional de Niños. Caja Costarricense de Seguro Social *fruizsalazar@gmail.com
Fecha recibido: 05 de noviembre de 2013 Fecha aceptado: 24 de abril de 2014

{kind=link}