








Serviços Personalizados
Journal
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkActa Médica Costarricense
versão On-line ISSN 0001-6002versão impressa ISSN 0001-6012
Acta méd. costarric vol.43 no.4 San José Out. 2001
Resumen
La Distrofia Miotónica es una enfermedad multisistémica de herencia autosómica dominante. El defecto molecular es una expansión del trinucleótido CTG presente en la región 3' no codificante (3' UTR) del gen DMPK, localizado en el cromosoma 19q13.3. El objetivo del estudio fue implementar el diagnóstico molecular de la Dm con el fin de contribuir a mejorar el manejo clínico de los pacientes afectados y a que el consejo genético sea más certero y preciso. El estudio se realizó en pacientes con diagnóstico mediante el uso de técnicas molecular, a saber, la hidridación de Southern y la PCR. Se obtuvo el diagnóstico molecular de 84 pacientes de 21 diferentes familias; en 34 se diagnosticó el defecto molecular. En 21 personas de familias donde se comprobó que segrega la mutación, el gen resultó normal. En 29 pacientes de ocho familias no se encontró la mutación. Se observó una correlación positiva entre la severidad de la enfermedad y el número de repeticiones CTG. Aquellos casos que resultaron negativos probablemente sean pacientes con mutaciones en otros genes, ya sea PROMM/DM2 u otras miotonias hereditarias. El diagnóstico molecular debe usarse como herramienta para logra la clasificación clínica de los pacientes. El abordaje correcto de la enfermedad, debido a que todavía no existe tratamiento, debe incluir, además del manejo clínico interdisciplinario, la prevención mediante el consejo genético basado en el diagnóstico molecular preciso de la condición de portador o portadora.
Descriptores: Distrofia miotónica, mutaciones instables, anticipación genética, Costa Rica, Hidridación de Southern, PCR, PROMM, consejo genético.
Recibido: 16 de julio de 2001.
Aceptado: 23 de octubre de 2001.
La Distrofia miotónica (DM) es una enfermedad neuromuscular degenerativa y progresiva que constituye la forma más común de distrofia muscular en adultos. Su incidencia varía en distintas poblaciones, para los japoneses se estima en 1/20.000, para caucásicos 1/8.000, llegando a 1/475 en ciertas regiones de Canadá. 1 Sin embargo, es extremadamente rara en africanos negros, en donde sólo se ha descrito una familia nigeriana afectada. 2
La DM es una enfermedad multisistématica, caracterizada por miotonía, desgaste y debilidad muscular progresivos, calvicie frontal, cataratas, problemas respiratorios, hipogonadismo, arritmias cardíacas producidas por defectos en el sistema de conducción del músculo cardíaco, resistente a la insulina y atrofia testicular. Se presenta generalmente en la tercera o cuarta década de la vida, pero puede ocurrir congénitamente presentándose de una manera más severa, incluyendo hipotonía, diplejia facial, retardo mental, defectos en la succipón y deglución. La forma congénita presenta alta tasa de mortalidad perinatal y aquellos pacientes que sobreviven a éste período desarrollan el cuadro clínico clásico de la enfermedad aproximadamente a los 10 años de edad .3
La DM presenta un patrón de herencia autosómico dominante con expresión variable y penetrancia incompleta. La variabilidad fenotípica de la enfermedad comprende desde individuos con expresión tardía leve que se manifiesta en cataratas, hasta individuos que presentan la forma congénita con expresión multisistémica. Además representa el ejemplo más contundente del fenómeno de anticipación genética debido a que, conforme se hereda ña mutación a través de las generaciones, el cuadro clínico de la enfermedad se manifiesta a una edad más temprana y de una manera más severa.
El defecto genético es una expansión del trinucleótido CTG en la región 3' no codificante (3' UTR) del gen DMPK (proteína quinasa de la distrofia miotónica por su similaridad de secuencia con la familia de proteína quinasa serina/treonina, las cuales se relacionan estrechamente con las proteínas quinasas dependientes de AMPc) localizada en el cromosoma 19q13.3, 4-8. La expansión es inestable, es decir, que conforme transcurren las divisiones celulares el número de repeticiones cambia, con gran cantidad de repeticiones en los pacientes con DM. La población normal presenta alelos entre 5 a 37 repeticiones mientras que los individuos afectados tienen de 50 a más de 1000 repeticiones. La mutación DM está presente en más de 98% de la población de pacientes con el cuadro clínico de DM estudiada hasta ahora, incluyendo diversos grupos étnicos en Europa, Asia, Australia, Norte y Sur América. 2,9
El gen se expresa principalmente en el músculo liso, esquelético (unión neuromuscular) y cardíaco (discos intercalados de los miocitos cardíacos y fibras de Purkinje), y en niveles más bajos también en el cerebro y en tejidos endocrinos. Existe un paralelo entre el patrón de expresión del gen en los diferentes tejidos y la forma fenotípica en que se manifiesta la enfermedad en los pacientes. 10
Por lo general, el fenotipo más severo está acompañado de los alelos con más repeticiones. Los individuos con expansiones pequeñas son mínimamente afectados, algunos presentan cataratas únicamente, mientras que otros son clínicamente normales. Con base en esto se han establecido varias categorías clínicas.
I.- Asintomática
II.- Leve: expresión de cataratas en adultos mayores de 40 años. En algunos casos se presenta debilidad y desgaste muscular leve.
III.- Clásica del adulto: se expresan los cambios distróficos y los rasgos neuromusculares típicos con miotonía al examen físico o electromiográfico en la adolescencia o temprano en la vida adulta.
IV.- Pediátrica: expresión < 10, los rasgos predominantes son retardo en el desarrollo y debilidad leve.
V.- Congénita: problemas neuromusculares, usualmente severos, claramente documentados al nacimiento. 11
No obstante, la presencia de esta correlación positiva, hay in traslape entre la longitud del alelo y al categoría clínica, lo cual indica que el tamaño de la amplificación no es suficiente para predecir con certeza la severidad y forma de expresión de la enfermedad. 12
El mecanismo molecular exacto por el cual ocurre la expansión es desconocido, al igual que el mecanismo fisiopatológico para la degenaración multisistémica 13 y aunque su base genética ya se ja dilucidado, muy poco se sabe sobre el defecto celular responsable de las manifestaciones pleiotrópicas de la enfermedad. 14-17 Sin embargo se han encontrado niveles cuantitativamente anormales del ARNm del gen DMPK y de la miotonima (nombre de la DMPK) en pacientes con DM.18,19
La DM no se ha estudiado en Costa Rica, no se conoce su prevalencia, ni se hace diagnóstico molecular a las familias afectadas. Por ser una enfermedad neurodegenetiva, en algunos casos severa e incapacitante, es de suma importancia conocer la población costarricense que porta la mutación, ya que ésta es la mejor manera de ofrecer un consejo genético adecuado. Es importante que en el país se comiencen a estudiar estas enfermedades y a implementar su diagnóstico. La presente investigación tiene como objetivo principal utilizar la biología molecular para determinar el tamaño exacto de la mutación en los pacientes afectados por DM y sus familiares, con el fin de evaluar la importancia de esta enfermedad como genético. Gracias a este estudio, se podrá confirmar el diagnóstico directo de la mutación por procedimientos moleculares y correlacionar la longitud de la repetición y la severidad de los síntomas clínicos. El inicio de estos estudios, permitirá estimar la prevalencia de este tipo de problemas neurológico en el país.
Materiales y Métodos
Población: Se estudiaron 84 pacientes con un diagnóstico clínico inicial de distrofia miotónica y sus familiares, pertenecientes a 21 familias diferentes. Los casos índices de las primeras familias con DM se obtuvieron de los registros presentes en el Hospital Nacional de Niños (H.N.N). Las familias restantes fueron captadas en la consulta externa del Hospital San Juan de Dios (H.S.J.D). El diagnóstico clínico de al menos un paciente por familia fue confirmado electromiográficamente. Los pacientes fueron contactados para invitarlos a participar en el estudio, fueron informados en detalle sobre las características de la enfermedad y las particularidades de su herencia. Aquellas personas que accedieron participar firmaron una fórmula de consentimiento informado aprobada por el Comité ético- Científico de la Universidad de Costa Rica.
Diagnóstico Clínico: El diagnóstico clínico se estableció después de la realización de un examen físico y electromiográfico. El examen físico incluyó la presencia o ausencia de cataratas, calvicie frontal , miotonía, problemas respiratorios y el grado de debilidad muscular. Para la electromiografía se evaluaron los músculos: abductor pulgar derecho y deltoides izquierdo, en donde se anotó el patrón miotónico, miopático, reclutamiento facial, baja amplitud y potenciales polifásicos, además se estudió la neurografia motora del miembro superior e inferior. Para efectos del análisis de los resultados se utilizó la clasificación clínica mencionada en la introducción, 11 debido a que los cuadros clínicos presentan una gran variabilidad individual, lo que hace imposible una descripción clínica detallada de cada paciente.
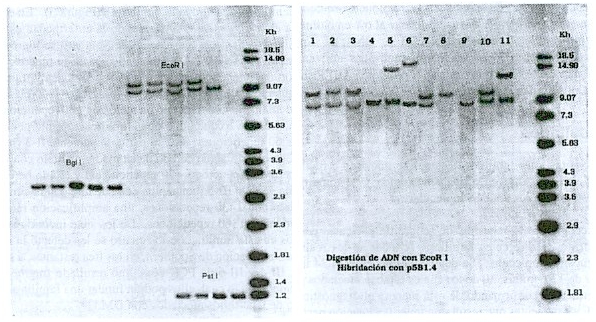
Diagnóstico Molecular: el ADN fue extraído de leucocitos de sangre periférica con fenol- cloroformo siguiendo los procedimientos usuales. 20 Las reacciones de digestión completa del ADN con las enzimas de restricción se hicieron de acuerdo con las especificaciones del fabricante. En general, 6mg de ADN fueron digeridos se separaron en geles de agarosa al 0.7% en buffer TBE 1X (tris-base, ácido bórico, EDTA). Los que fueron transferidos a una menbrana de nylon por el método de Southern (transferencia del ADN a un soporte sólido). Los filtros se hidridaron toda la noche con las sondas específicas (segmento de ADN complementario a una región de un gen) para la mutación DM: la sonda p5B1.4 que detecta fragmentos normales de 8.6 Kb y 9.8 Kb previa digestión con la enzima Eco RI y de 3.4 Kb cuando se utiliza al BgII; y la sonda pM10M6 que detecta un fragmento normal de 1.2 Kb con la enzima PstI. Las sondas fueron marcadas con fósforo radiactivo. Las membranas se lavaron en SSC y SDS y se expusieron a una película de Rayos X que permaneció a – 70° C entre 5-7 días. 21 Después de reveladas las películas, se estimó el tamaño de la amplificación en todos los individuos analizados por comparación con el marcador de peso molecular (Figura 1).
La reacción en cadena de la polimerasa (PCR, amplificación de un segmento específico del ADN) se realizó en aquellas muestras que presentaron fragmentos normales mediante el procedimiento descrito anteriormente. Cada muestra fue digerida con Hind III por 1h a 37° C; según lo establece el protocolo de Monckton et al. 22 Luego se amplificaron 100 ng siguiendo las condiciones usuales, usando buffer 11X, 0.5 unidades de Biotaq polimerasa, agua, los iniciadores DM-A (5-'CAGTTCACAACCGCTCCGAGC-3') y DM-BR (5'CGTGGAGGATGGAACACGGGAC-3') los cuales producen un fragmento de 252 pares de bases en las muestras sin repeticiones. El volumen final de reacción fue de 10 ml. El perfil utilizado en el termociclador T3 Biometra fue: 28x (96°C por 45", 68 °C por 45", 70°C por 3'), 68°C por 452 y un periodo de extensión final a 72° C por 10'.
Los productos de la PCR se corrieron en un gel de agarosa al 1% en buffer TBE 0.5X toda la noche a 300V a 4°C. se transfirió el ADN a una membrana y se realizó una hibridación utilizando una sonda consistente de un fragmento con 52 repeticiones CTG. La sonda fue marcada con a-32P-dCTP. La membrana se hibridó toda la noche, posteriormente fueron lavadas en SDS y SSC y expuestos a una película de rayos X toda la noche. El tamaño de los fragmentos se determinó utilizando el Software Kodak 1D22 (Figura 2a).
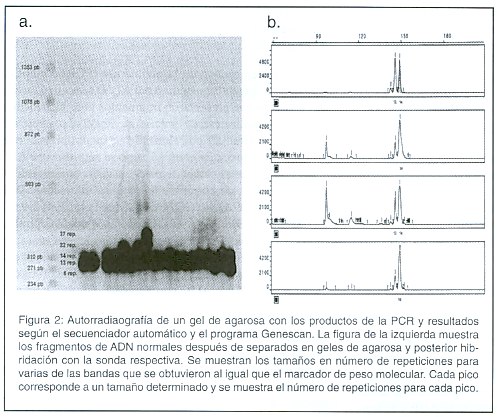
Los resultados de la PCR se confirmaron utilizando un secuenciador automático. Para esto, se utilizaron los iniciadores: DM-CF-5'-AACGGGGCTCGAAGGGTCCT-3'y DM-DR-5'- CAGGCCTGCAGTTTGCCCATC-3', uno de los cuales estaba marcado con fluorescencia. El producto de la PCR en este caso es un fragmento de 106 pares de bases cuando no hay repeticiones. Los productos se corrieron en un gel de acrilamida desnaturalizante al 6% en buffer TBE 1X utilizando el secuenciador 373 A DNA de Applied Biosystem. Las muestras fueron analizadas utilizando el Software Genescan Analysis con el programa ABI Pism 373 XL Collection (Figura 2b).
Resultados
Las visitas a las familias afectadas se iniciaron a finales de 1997, se construyó el árbol genealógico y se tomaron las muestras para los análisis moleculares.
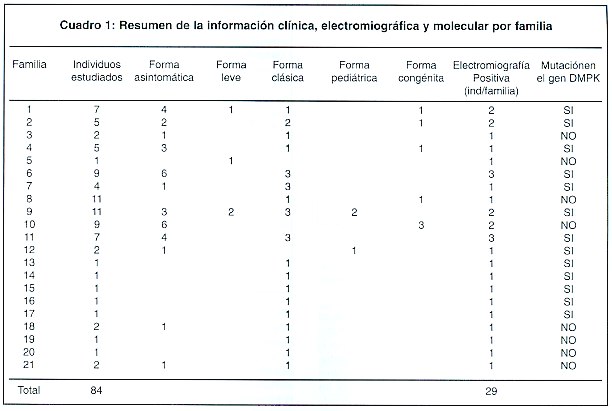
Se recogieron 84 muestras de sangre de pacientes pertenecientes a 21 diferentes familias. A todas se les realizó el diagnóstico molecular. Se detectó la mutación en 34 individuos de 13 familias, 30 de los cuales estaban afectados por la enfermedad, confirmando de esta manera el diagnóstico clínico, y 4 pacientes que resultaron tener la mutación permanecen asintomáticos hasta el momento, 21 personas de las mismas 13 familias no heredaron la mutación. Seis pacientes afectados de 13 familias, uno con la forma pediátrica, dos con la congénita y tres con la clásica no se estudiaron en el laboratorio por dificultades en la toma de la muestra de sangre. Por lo tanto, los pacientes sintomáticos suman un total de 36.
El estudio molecular determinó que 29 individuos, entre afectados y familiares, pertenecientes a otras ocho familias, que habían sido clínicamente diagnosticadas como DM, no son portadores de la mutación en el gen DMPK. El Cuadro 1 presenta una descripción detallada de cada familia y de los casos afectados. Dos de esas familias estaban en los registros iniciales tomados en el Hospital Nacional de Niños y las otras 6 fueron apareciendo conforme la investigación iba avanzando.
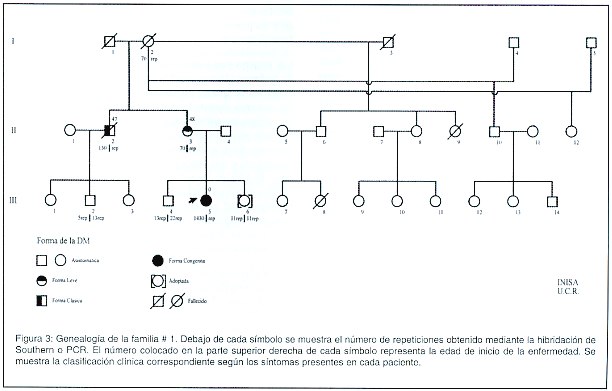
En las 7 familias diagnosticadas con DM y en las que se cuenta con la genealogía, el número de afectados varía de una a otra, al igual que la clasificación clínica, es decir, en unas familias se presenta únicamente dos formas (clásica y congénita) mientras que en otras se presentan tres forma (leve, clásica y pediátrica). También hay diferencias entre el sexo que está transmitiendo la enfermedad, en algunas familias es sólo el sexo femenino, en otra sólo el sexo masculino, y en otras los dos sexos. Sin embargo, todas tienen dos cosas en común: a- en todas las familias el número de repeticiones CTG aumenta de una generación a la siguiente; b- la severidad de la enfermedad también aumenta de una generación a la otra. Un ejemplo lo constituye la familia #1 (Figura 3). En esta familia el sexo que está transmitiendo la enfermedad es el femenino. Mediante la hibridación de Southern se logró determinar que la abuela I-2 (asintomática hasta su muerte, 75 años) portaba una amplificación de 70 repeticiones y transmitió la mutación a dos de sus hijos. El individuo II-2 (clásica, edad de inicio: 47 años) recibió un alelo con 130 repeticiones de su madre, con lo que hubo una amplificación intergeneracional de 60 repeticiones. Por su parte, la paciente II-3 (leve, edad de inicio: 48 años) heredó de su madre un alelo con igual número de repeticiones (70 repeticiones) y ella le heredó a una hija, la niña III-5 (congénita, edad de inicio al nacimiento) un alelo con 1430 repeticiones, una amplificación intergeneracional de 1360 repeticiones. De los siete individuos analizados en esta familia, a estos cuatro se les detectó la mutación por hibridación de Southern, en los tres restantes, a saber, III-2, III-4, III-6, la PCR dio como resultado fragmentos normales, con lo cual, ellos podrán fundar una familia sin el riesgo de tener hijos afectados con DM.
Como se puede observar en la Figura 3, la edad de manifestación disminuye a través de las generaciones, lo cual correlaciona con el incremento en el tamaño de la mutación. También se logra observar un incremento en la severidad del cuadro clínico de los pacientes conforme transcurren las generaciones.
En el cuadro II se aprecia claramente como correlaciona positivamente la severidad de la enfermedad y el tamaño de la mutación, además se muestra la distribución de los casos afectados por categoría clínica.
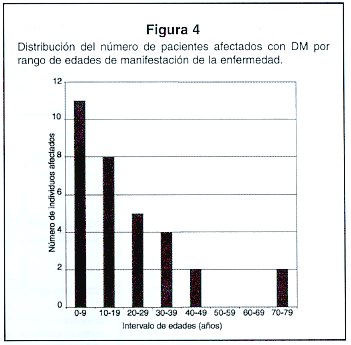
Una distribución del número de casos que se presentaron en este estudio por rango de edades a las cuales dichos pacientes empezaron a desarrollar síntomas de la DM se presenta en la Figura 4. Como se observa, la mayoría de los casos estudiados y a los cuales se les realizó el análisis molecular, iniciaron el cuadro clínico antes de los veinte años, no habiéndose encontrado a ningún paciente entre los cincuenta y sesenta y nueve años.

Discusión
La presente investigación logró obtener información molecular de 84 individuos en 21 familias costarricenses que tenían un diagnóstico clínico de DM. Solo 13 de éstas familias tienen la mutación en el gen DMPK, que causa la DM tipo 1. Por el momento, el análisis molecular ha confirmado el diagnóstico clínico de DM en 30 pacientes clínicamente afectados. Existen también otras seis personas afectadas clínicamente en las familias donde se confirmó la presencia de la mutación, y otros cuatro individuos portadores asintomáticos, por lo que hasta el momento hay al menos 40 individuos portadores del alelo con amplificación CTG en Costa Rica. Este estudio ha logrado contribuir a la correcta clasificación clínica de 21 familias, quedando todavía por analizar a nivel molecular al menos otras 32, ya que hasta la fecha sabemos que en Costa Rica existen al menos 53 familias con diagnóstico clínico inicial de DM.
Este es el primer y único estudio que se ha llevado a cabo en Costa Rica sobre la DM que incluye el análisis molecular; aunque todavía esta en sus inicios, es evidente que esta enfermedad emerge como una patología importante de prevenir debido a la gran carga que representa para las familias y la sociedad. Sería conveniente que su estudio continúe, debido a que la DM es uno de los padecimientos hereditarios que están contribuyendo a la morbilidad y mortalidad de la población costarricense, al igual que en otros países, donde se ha observado que la esperanza y la calidad de vida se ve reducida en los pacientes afectados. Un gran porcentaje de los recién nacidos con la forma congénita mueren durante el primer año de vida, los que sobreviven muestran el cuadro clínico de la forma clásica a la edad de 10 años, muchos de ellos deben ser sometidos a intervenciones quirúrgicas para corregir sus deformaciones esqueléticas, 23 algunos muestran retardo mental y un porcentaje debe venir confinando a la silla de ruedas. La esperanza de vida promedio de éstos niños es de 35 años. 24
Las mujeres con tamaños de amplificación dentro del rango de la forma clásica, muchas veces están asintomáticas en el momento de quedar embarazadas y comienzan a manifestar los síntomas durante la gestación 25. Estos embarazos son de alto riesgo, muestran atonía uterina, placenta previa, placenta retenida, polihidramnios, abortos espontáneos, mortinatos sufrimiento fetal, arritmia cardíaca materna, sensibilidad a los anestésicos y relajantes y depresión respiratoria postoperatoria. 26 EL 12% de los niños nacen muertos o mueren al nacer, el 9% sobreviven afectados gravemente, y el 29% manifiestan la enfermedad posteriormente. Por estas razones se debe ofrecer el diagnóstico molecular predictivo y presintomático a las mujeres de familias con DM con el fin de adelantarse a las complicaciones obstétricas 27.
Un estudio longitudinal de 180 pacientes afectados por la forma clásica e Holanda demostró que la esperanza de vida se ve reducida, a los 45 años han sobrevivido un 88% de los pacientes, un 7% menos que la población general, a los 65 años solamente ha sobrevivido un 18% de los afectados en comparación con el 78% de la población general. Un 73% de las muertes ocurrieron por neumonías y arritmias cardíacas, causas directamente atribuibles a la distrofia miotónica. Un 50% de los pacientes estaban en sillas de ruedas cuando fallecieron. 24
La prevalencia de la DM en Costa Rica es desconocida, pues se ignora el número exacto de familias y el número total de afectados con la enfermedad. Sin embargo, tomando en cuenta la prevalencia de DM en caucásicos es ed1/8000, que en africanos y amerindios es casi nula 28 y considerando que la población costarricense es producto de una mezcla racial, en donde el componente genético europeo es el más importante de las tres etnias originales (Español, Indígena y Africano), se podría esperar que la prevalencia en Costa Rica sea similar a la de caucásicos, aunque un poco menor debido a esa mezcla de la que somos producto.
De las 21 familias con un diagnostico molecular, ocho no tienen la amplificación, aunque habían sido inicialmente diagnosticadas como DM. Se ha descrito que aproximadamente un 1-2% de los pacientes que presentan el cuadro clínico no presentan la amplificación en el gen DMPK.8 Thornton et al 29 fueron los primeros es describir los primeros tres casos de DM sin la amplificación CTG. El diagnóstico molecular, por lo que ellos concluyeron que sus tres pacientes podrían presentar una condición distinta a la DM. Esa condición, que es muy similar a la DM, es la Miopatía Miotónica Proximal (PROMM); caracterizada por presentar miotonía, debilidad facial, debilidad distal y proximal, cataratas, calvicie, problemas cardíacos, las cuales, son también las principales manifestaciones clínicas en la DM. Sin embargo, el cuadro clínico en PROMM es más leve y el fenómeno de anticipación no es tan claro como en la DM. Hasta la fecha no se ha descrito un caso congénito de PROMM. 30-32 Recientemente Ranum et al., 31 reportaron una familia con una condición muy similar a DM a la que llamaron DM tipo 2 (DM2), por presentar las mismas manifestaciones clínicas de la DM pero que no mostraban la mutación en el gen DMPK. Ellos lograron localizar el gen de la enfermedad en esta familia en una región del brazo largo del cromosoma 3. Sin embargo, más recientemente Ricker et al. 33 mostraron por primera vez que el locus de la enfermedad para PROMM estaba ligado al locus recién descrito para DM2 en el brazo largo del cromosoma 3. Ellos mencionan que debido a que los fenotipos clínicos de PROMM y DM2 parecen traslaparse, las dos enfermedades podrían ser causadas por la misma mutación, ser condiciones alélicas o ser causadas por genes ligados con funciones relacionadas.
La ausencia de la mutación DM en individuos con diagnóstico clínico, permite sospechar que sean pacientes con PROMM, DM2 o también podrían ser pacientes que están siendo afectados por otras miotonías hereditarias, pues, vale la pena aclarar que hay un número de enfermedades que tiene miotonía como uno de los signos principales, a saber, paramiotonía congénita, miotonía de Thomsen, miotonía congénita, entre otras. 34 Esta situación es la que podría estarse presentando en alguna de estas ocho familias de Costa Rica, algo que todavía queda por estudiarse y comprobarse con análisis moleculares.
La DM es una de las enfermedades de mayor variabilidad fenotípica que se conocen, encontrándose desde n gravemente afectados, hasta individuos en los que las cataratas es el único síntoma apreciable. Por lo general, los pacientes gravemente afectados presentan un número de repeticiones CTG relativamente grande, mientras que individuos con cataratas presentan un número de repeticiones relativamente pequeño. Sin embargo, existe un traslape entre la severidad de la enfermedad y el tamaño de la mutación (Cuadro II), por lo que el tamaño de la mutación no debe ser tomado como único factor para determinar la forma y la severidad de la enfermedad. Esto concuerda con lo descrito por Lavedan et al. 12
La mayoría de la población afectada y estudiada a nivel molecular hasta el momento (N=19) en nuestro país están dentro del rango de edades entre 0 y 19 años. Los restantes 13 casos están den el ámbito de edades entre 20 y 79 años. Como consecuencia de la inestabilidad meiótica, característica de la mutación DM y de su forma de transmisión, las personas con edades entre 0 a 19 años son las que presentan el cuadro clínico más severo. En este sentido, el consejo genético basado en el examen molecular podría ayudar a las familias donde se transmite la mutación DM, a identificar los portadores con alto riesgo de procrear niños afectados con la forma pediátrica o congénita, con el consecuente beneficio a largo plazo al disminuir el número de niños afectados por una enfermedad de lato costo emocional y económico para las familias y la sociedad. Hay que aclarar que el consejo genético debe ser utilizado como una herramienta para ayudar a las parejas a tomar las mejores decisiones en el momento de establecer una familia, decisiones que se toman de acuerdo con los valores y principios éticos de cada persona.
El diagnóstico molecular de la DM es una realidad en Costa Rica en estos momentos. La información molecular que se obtiene ahora puede ser usada para brindar un asesoramiento genético más seguro y confiable a las personas portadoras de la mutación y con riesgo de transmitirla a sus hijos. Debido a que las miopatías constituyen un grupo muy grande de enfermedades, la biología molecular es una herramienta muy valiosa para confirmar o descartar un diagnóstico equivocado, como la situación encontrada en las ocho familias reportadas en esta investigación. Por lo tanto, el diagnóstico clínico debería apoyarse siempre en la biología molecular, para lograr la reclasificación clínica e ir desarrollando conciencia entre los clínicos sobre la importancia de establecer un diagnostico correcto, con fines de prevención. Tomando en consideración que el diagnóstico molecular de la DM es ahora posible, sería conveniente desarrollar un plan de acción multidisciplinario, que permita el seguimiento adecuado de los pacientes y que favorezca el desarrollo de la investigación en el estudio de las enfermedades que presentan miotonía como síntoma principal en la población costarricense.
Abstract
Myotonic dystrophy (MD) is a multisystemic disease wit an autosomal dominant inheritance. The genetic defect is an unstable mutation due to the expansion of the triple CTG in the 3' unstranslated region (3' UTR) of the DMPK gene on chromosome 19q13.3. the objective of this work was to implement the molecular diagnosis of the disease in order to improve the clinical management and the genetic counseling offered to patients with clinical diagnosis of Dm and their relatives. Two technical diagnostic procedures were used, Southern blot and PCR for confirming the clinical diagnosis. We studied 84 members from 21 different families, obtaining the following results: 21 asymptomatic individuals without the mutation (normal), 34 have the mutation (4 of the are still asympomatic) and 29 patients in 8 families with a previous clinical diagnosis of DM do not have the DM mutation. The size of the mutation in positively correlated with severity of symptoms. Those cases with no DM mutation probably correspond to other mutations, such as PROMM/DM2 or other inherited myotonies. Molecular diagnosis must be used as a tool for propering a clinical classification of the patients. The adequate clinical approach, since there is no treatment so far, should include, besides a multidisciplinary clinical management, prevention through genetic counseling based on the exact molecular diagnosis of the carriers.
Agradecimientos y colaboradores
A la Vicerrectoría de Investigación de la Universidad de Costa Rica por el financiamiento del proyecto N° 742-98-322. Al Organismo Internacional de Energía Atómica por crear la infraestructura para el estudio de enfermedades causadas por mutaciones inestables. Al Ministerio de Ciencia y Tecnología, al CONICIT y FUNDEVI por su apoyo económico. A los doctores Sandra Silva y Carlos de Céspedes por su apoyo durante la realización de este trabajo, y por la revisión del primer manuscrito. Al Grupo de Investigación en Mutaciones Dinámicas de la Universidad de Glasgow, en especial al Prof. Keith Johnson y Darren Monckton por su asesoría y ayuda.
Abreviaturas: 3' UTR, región 3' no codificante de un gen; ADN, Ácido desoxirribonucleico; AMPc, Monofosfato cíclico de adenosina; ARNm, Acido ribonucleico mensajero; CENARE, Centro Nacional de Rehabilitación; CTG, Citosina, timina, guanina; DM, Distrofia miotónica; DM2; Distrofia miotónica tipo 2; DMPK, Proteína quinasa de la distrofía miotónica; H.N.N., Hospital Nacional de Niños; HS.JD. Hospital San Juan de Dios, INIS, Instituto de Investigaciones en salud; PCR, Reacción en cadena de la polimersa; PROMM, Miopatía miotónica proximal; UCR, Universidad de Costa Rica.
Referencias
1. Zerylnick C, Torroni A, Sherrnan SL, Warren ST. Normal variation at the myotonic dystrophy locus in global human populations. Am J Hum Genet 1995; 56: 123-130. [ Links ]
2. Krahe R, Eckhart M, Ogunniyi A O, Osuntokun B O, Siciliano M, Ashizawa T. De novo myotonic dystrophy mutation in a Nigerian kindred. Am J Hum Genet 1995; 56: 1067-1074.
3. Harris S, Moncrieff C, Johnson K. Myotonic dystrophy: will the real gene please step forward. Hum Mal Genet 1996; 5: 1417-1423. [ Links ]
4. Aslanidis C, Jansen G, Amerniya C, Shutler G, Mahadevan M, Tsilfidis C, et al. Cloning of the essential myotonic dystrophy region and mapping ofthe putative defect. Nature 1992; 355 (6): 548-551.
5. Buxton J, Shelboume P, Davies J, Jones C, Van Tongeren T, Aslanidis C, et al. Detection of an unstable fragment of DNA specific to individuals with DM. Nature 1992; 355: 547-548.
6. Fu Y H, Pizzuti A, Fenwick R G, King J Jr, Rajnarayan S, Dunne O W, et al. An unstable triplet repeat in a gene related to myotonic muscular distrophy. Science 1992; 255: 1256-1258.
7. Harley H, Brook J. D, Rundle S, Crow S, Reardon W, Buckler A, et al. Expansion of an unstable DNA region and phenotypic variation in DM. Nature 1992; 355: 545-546.
8. Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic distrophy mutation: An unstable CTG repeat in the 3' untranslated region of the gene. Science 1992; 255: 1253-1255.
9. Meiner A, Wolf C, Carey N, Okitsu A, Johnson K, Shelboume P, et al. Direct molecular analysis of myotonic dystrophy in the gerrnan population: important considerations in genetic counselling. J Med Genet 1995; 32: 645-649.
10. Jansen G, Willems P, Coerwinkel M, Nillesen W, Smeets H, Vits L, et al. Gonosomal mosaicism in DM patients: lnvolvement of mitotic events in (CTG)n repeat variation and selection against extreme expansion in sperrn. Am J Hum Genet 1994; 54: 575-585.
11. Hecht B K, Donnelly A, Gedeon A K, Byard R W, Haan E A, Mulley J C. Direct molecular diagnosis of the myotonic dystrophy. Clin Genet 1993; 43: 276-285.
12. Lavedan C, Hofmann-Radvanyi H, Shellboume P, Rabes J, Duros C, Savoi D, et al. Myotonic dystrophy: size and sex-dependent dynarnics of CTG meiotic instability, and somatic mosaicism. Am J Hum Genet 1993; 52: 875-883.
13. Thornton C, Wymer P J, Simmons Z, McClain C, Moxley ID R Expansion of the myotonic dystrophy CTG repeat reduces expresion of the flanking DMAHP gene. Nat Genet 1997; 16: 407-409.
14. Caskey C T, Swanson M S, Timchenko L T. Myotonic dystrophy: discussion of the molecular mechanism. Cold. Spring. Harb. Symp. Quant Biol 1996; 61: 607-613. [ Links ]
15. Johnson K J, Boucher C A, King S K, Winchester CL, Bailey M E S, Hamilton G M, et al. ls myotonic dystrophy a single-gene disorder? Biochem Soc Trans 1996; 24: 510-513.
16. Bhagwati S, Leung B, Shaquif S, Ghatpande A. Myotonic dystrophy: decreased levels of myotonin protein kinase (Mt - PK) leads to apoptosis in muscle cells. Exper Neurology 1997; 146: 277- 281.
17. Chahine M, George Jr LA. Myotonic dystrophy kinase modulates skeletal muscle but not cardiac voltage-gated sodium chanels. FEBS Letters 1997; 412: 621-624. [ Links ]
18. Taneja KL, McCurrach M, Schalling M, Housman D, Singer R Foci of trinucleotide repeat tanscripts in nuclei of DM cells and tissues. J Cell Bio11995; 128: 995-1002.
19. lshii S, Nishio T, Sunohara N, Yoshihara T, Takemura K, Hikiji K, et al. Small increase in triplet lenght of cerebellum from patients with myotonic dystrophy. Hum Genet 1996; 98: 138-140.
20. Sambrook J, Fritsch E. F & Maniatis T. Molecular cloning. A laboratory manual. 2da Ed. New York: Cold Sping Harbor Laboratory Press, 1989.
21. Shelboume, F., Winqvist, R, Kunert, E., Davies, J., Leisti, J., Thiele, H., et al. Unstable DNA may be responsible for the incomplete penetrance of the myotonic distrophy phenotype. Hum Mal Gen 1992; 1: 467-473.
22. Monckton D, Wong L. J., Ashizawa T, & Caskey T. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analises. Hum Mol Genet 1995; 4 (1): 1-8.
23. Harper P. Myotonic Dystrophy as a Trinucleotide Repeat Disorder-A Clinical Perspective. En: Wells R, Warren S, ed. California: Academic Press. 1998: 115-130. [ Links ]
24. Die-Smulders C, Howeler C, Thijs C, Mirandolle J, Anten H, Smeets H, et al. Age and causes of death in adult-onset myotonic dystrophy. Brain 1998; 121: 1557-1563.
25. Keriakos R, Aziz & Sidra L. Myotonic dystrophy in pregnancy. Journal of Obstetrics and Gynecology 1999; 19 (1): 71-73. [ Links ]
26. Golbus M, Simpson J. Genetics in Obstetrics & Gynecology. 2da ed. México: Saunders, 1992. [ Links ]
27. Milunsky A. Genetic disorders and the fetus. 3ra ed. Baltimore: The Johns Hopkins University Press, 1992. [ Links ]
28. Deka R, Majumder P, Shriver M, Stiver D, Zhong y, Yu M L, et al. Distribution and evolution of CTG repeats at the myotonin protein kinase gene in human populations. Genome Res 1996; 6: 142-154.
29. Thornton C, Griggs R, Moxley R. Myotonic Dystrophy with no trinucleotide repeat expansion. Ann Neurol 1994; 35: 269-272. [ Links ]
30. Abbruzzesse C, Krahe R, Liguori M, Tessarolo D, Siciliano M, Ashizawa T, et al. Myotonic dystrophy phenotipe wihtout expansion of (CGT)n Repeat: an entity distinct from proxomal miotonic myopathy (PROMM)? J Neurol 1996; 243: 715-721.
31. Rannum L, Rasmussen P, Benzow K, Koob DM, Day WJ. Genetic mapping of a second myotonic distrophy locus. Nat Genet 1998; 19: 196-198.
32. Ricker K. Myotonic dystrophy and proximal miotonic myopathy. J Neuro11999; 146: 334-338.
33. Ricker K, Grimm T, Koch MC, Schneider C, Kress W, Reimers CD, et al. Linkage of proximal myotonic myopathy to chomosome 3q. Neurology 1999; 59: 170-171.
34. Koty P, Pegoraro E, Hobson G, Marks HG, Turel A, Flagler D, et al. Myotonia and the muscle chloride channel: dominant mutations show variable penetrance and founder effect. Neurology 1996; 47: 963-968.
1 Instituto de Investigaciones en Salud, Universidad de Costa Rica.
2 Escuela de Biología, Universidad de Costa Rica.
3 Servicio de Neurología, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera"
4 Servicio de Neurología, Hospital San Juan de Dios.
Correspondencia: Fernando Morales Montero. INISA, UCR. Ciudad Universitaria Rodrigo Facio, San José, Costa Rica. Correo electrónico: famorale@cariari.ucr.ac.cr