










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Costarricense
versión On-line ISSN 0001-6002versión impresa ISSN 0001-6012
Acta méd. costarric vol.43 no.3 San José jul. 2001
Cardiopatías Congénitas en Adolescentes y Adultos
Análisis de los pacientes egresados de los hospitales generales del área metropolitana, durante 1998
Patricia Hernández-Cordero,1 Carlos Salazar-Vargas 2
Resumen: La incidencia y la prevalencia de las cardiopatías congénitas (CC) en nuestro medio y en otros países en vías de desarrollo son desconocidas. A pesar de que contamos con una extensa cobertura en salud, vemos con frecuencia adultos no tratados con este tipo de padecimientos. Debido al crecimiento demográfico y a la disminución de la mortalidad infantil el médico actual se enfrenta a más enfermos con CC, a los cuales debe diagnosticar, referir para tratamiento y aconsejar. Con el objeto de conocer más de la realidad nacional específica en este campo y a la edad en el momento del diagnóstico estudiamos todos los pacientes egresados de los Hospitales Generales del área metropolitana con CC, ya que a éstos son referidos desde todo el territorio nacional.
Se escogió el año 1998, para que la investigación fuera reciente y para observar al menos 2 años de seguimiento después del tratamiento. Hubo un total de 45 pacientes, 75% mujeres, y 25% hombres, con un rango de edad de 15 a 64 años. La mayoría habían nacido en San José. Las CC encontradas en orden de frecuencia fueron: CIA(66.6%), CIV (11.5%), Tetralogía de Fallot (4.4%), y un caso de cada una de las siguientes: coartacion de la aorta, estenosis aórtica, anomalía de Ebstein, tronco arterioso común, Bloqueo AV congénito y aneurisma roto del seno de Valsalva. En 61.53% de los enfermos el diagnóstico se hizo a edades mayores a los 10 años, y el 81% estaban entre 20 a 40 años. El examen definitivo más utilizado fue la ecocardiografía.
De las 34 mujeres, 12 tuvieron hijos, hubo 48 partos vaginales, y 8 cesáreas sin complicaciones. Una paciente con CIA tuvo un hijo con CIA.
Treinta y dos individuos fueron intervenidos durante el año en estudio, y todos sobrevivieron. La clase funcional (NYHA) postoperatoria esI en 30 de ellos. Otras razonesde internamiento a los hospitales fueron obstétricaso para tratamiento médico.
Veinticuatro pacientes trabajan normalmente, y dos son ya pensionados, nueve han tenido problemas en el desempeño de sus funciones, uno con Síndrome de Down y uno con tronco arterioso no trabaja, y en nueve no se pudo obtener este dato.
Como se puede deducir de los datos presentados, nosotros estamos diagnosticando tardíamente nuestros pacientes con CC comparados con otras latitudes. Una mayoría llega a los Hospitales metropolitanos con el diagnóstico hecho. También se ve que el tratamiento quirúrgico se realiza exitosamente y que los sujetos se reintegran en buen número a la fuerza laboral y procrean normalmente. El estudio sugiere que existe un número de enfermos que aún no consultan o no son descubiertos.
Recibido: 30 de marzo de 2001 Aceptado: 14 de agosto de 2001
En nuestro tiempo el médico está expuesto a un número cada vez mayor de individuos con malformaciones congénitas de toda índole, incluyendo por supuesto los cardiovasculares. 1 Esto puede deberse al crecimiento demográfico, a una disminución en la tasa de mortalidad infantil y a la prolongación del rango de vida de los adultos. 2
El incremento en el número de enfermos, demanda un mejor conocimiento de la anatomía, de la fisiología, de la manera de presentación y del tratamiento de cada anomalía o de cada cardiopatía específicamente, y tambien de las implicaciones psicológicas producidas por las malformaciones en las personas que las padecen. 3
Conocer la problemática de estas condiciones es muy importante, y el médico debe de manejarlas para poder aconsejar a sus pacientes. La mayoría de los adultosy adolescentes con cardiopatías congénitas (CC) tienen conocimiento sin suficientes sobre sus trastornos cardíacos. Al volverse responsable el adulto joven, debe reconocer en mayor grado sus problemasy limitaciones, sus eventuales complicaciones, la posibilidad de transmisión a su progenie, y en general tomar control de su salud.1
Los progresos en el diagnóstico y el tratamiento de estos pacientes, han mejorado su supervivencia de manera importante. Muchos adultos pueden tener anomalías silenciosas o poco sintomáticas, o haber sido acertadamente diagnosticados pero no sometidos antes a procedimientos correctivos por no requerirlo, o bien porque sus lesiones podían ser tratadas durante la vida adulta, eran ya inoperables o sencillamente porque escaparon a ser descubiertas tempranamente.1,4
Siendo el nuestro un país pequeño, que cuenta con una extensa cobertura sanitaria y con un adecuado sistema de comunicación la presencia de pacientes adultos con CC en nuestros servicios de hospitalización despierta interrogantes. Sin embargo el fenómeno del diagnóstico tardío o equivocado existe también en otras partes del mundo, por ejemplo, sólo en EEUU se estima que existe la nada despreciable cifra de alrededor de 50000 adultos con CC no reconocida, mal diagnosticada o ya detectada pero no tratada.5
Se supone que estas anomalías deberían haber sido descubiertasy tratadas en edades menores, aunque algunas CC son compatibles con largos períodos sin síntomas o con un estado funcional bastante cercano a lo normal.
El estudio de las CC al igual que de otras condiciones nosológicas, implica varios aspectos importantes, que van desde lo puramente estadístico a lo mas profundamente humano. En Costa Rica y en otros países en vías de desarrollo no existen datos recientes sobre la incidencia, la prevalencia y otras características de estas malformaciones en la población adulta.1,6
El objetivo principal de este trabajo es describir todos los adultos con CC, que egresaron de los Hospitales Generales del área metropolitana durante un año, cuáles fueron sus lesiones, cómo fueron diagnosticados y tratados, así como su evolución y su funcionalidad en nuestro esquema social.
Materiales y Métodos
Para realizar esta investigación, los pacientes con CC, se extrajeron del total de pacientes egresados desde el 1ero de enero hasta el 31 de diciembre de 1998 de losHospitales Generales del área metropolitana: Hospital Calderón Guardia (HCG), Hospital México (HM), y Hospital San Juan de Dios(HSJD), antes llamados de referencia,7 ya que sus áreas de atracción cubren todo el territorio nacional. Se escogieron los expedientes clínicos de lospacientescon CC, admitidos por cualquier motivo, fuera o no relacionado con su CC.
Se seleccionó el año de 1998, para obtener información más reciente y cercana al inicio de la investigación y también para contar por lo menos con dos años de seguimiento, sobre todo de quienes hubieren sido tratados quirúrgicamente.
Se presentó un documento exponiendo los objetivos de este estudio, ante los respectivos Comités de Investigación de dichos centros, basado en el "Instructivo para propuestas de Investigación del HM", el cual fue estudiado y eventualmente aprobado por éstos.8
Se solicitó la lista de pacientesa la Oficina de Bioestadística de cada nosocomio, utilizando los códigos correspondientes a las CC según la última versión de la "Clasificación Internacional de las Enfermedades" (CIE10) de la Organización Mundial de la Salud,9 todos los pacientes menoresde 12 años fueron excluidos.
Se elaboró un cuestionario para recoger la información de los expedientes clínicos. Las variables cuantitativas fueron: edad del paciente y edad en que se realizó el diagnóstico y las cualitativas fueron: el género, lugar de nacimiento y residencia de acuerdo a cada provincia de nuestro país, el centro de salud donde se realizó el diagnóstico, el método utilizado, los defectos asociados encontrados, el tipo de CC, y el tratamiento recibido. Si este fue médico o quirúrgico, el tipo de corrección, y la clase funcional basada en la clasificación de New York Heart Association (NYHA).10 Otras variables fueron la paridad, cómo fueron los partosy determinar si los hijos de estos individuos eran normales, por último se estudió el desempeño de los pacientes en su trabajo, para conocer sus limitaciones.
La clasificación de lasCC se basó en la expuesta por Friedman y Child en el texto de "Medicina Interna de Harrison".11
En los casos en que se pudo o fue necesario, el paciente o sus familiares fueron contactados telefónicamente, para completar el protocolo.
De acuerdo al tiempo, este estudio es retrospectivo, con respecto al período y secuencia es transversal. El análisisy alcance de los resultados se realizó en forma descriptiva.
Los resultados fueron analizados mediante el programa Statistical Package for the Social Sciences(SPSS).
Se usaron distribuciones de frecuencia, porcentajes, y medidas de tendencia central. La formula de Mcnemar se aplicó para determinar la efectividad del tratamiento quirúrgico en los pacientesque fueron intervenidos quirúrgicamente.12
El lugar de residencia se expresó a travésde tasas por 100000 habitantes, de acuerdo a la población de cada provincia en el año 1998.
Resultados
Durante 1998, 45 adultosy adolescentes con CC, fueron egresadosde los tres hospitales de referencia del área metropolitana por diferentes motivos, de ellos 34 (75.5%) eran mujeresy 11 (24.4%) eran hombres, para una proporción de 3:1. El rango de edad de las mujeres fue de 15 a 64 años, con un promedio de 30 añosy el rango para los hombresfue de 15 a 57 años, con un promedio de 26.6 años. Ningún enfermo falleció.
Tipo de cardiopatía congénita: Las CC encontradas según su frecuencia fueron: CIA en 30 individuos(66.6%), 24 de tipo ostiumsecundum, y 4 de tipo seno venoso, una estaba asociada a dextrocardia y otra se asoció a EP. En 7 pacientesse encontró CIV (11.5%), una asociada a PCA y 3 a EP. La TF se halló en 2 pacientes (4.4%; Cuadro 1).
Las siguientes cardiopatías se presentaron sólo una vez, para un porcentaje de 2.2% cada una: coartación de aorta (Co/A), estenosis pulmonar (EP), Bloqueo atrioventricular congénito (B/AV), Anomalía de Ebstein, estenosis aortica (E Ao), Aneurisma roto del seno de Valsalva, y tronco común (TC).
Edad en que se hizo el diagnóstico: Se pudo conocer con certeza la edad del diagnóstico en 39 individuos de todo el grupo o sea en un 86.6% de la muestra. Tres personas habían sido diagnosticadas cuando eran menores de un año (7.69%), 12 durante la infancia o sea de 1 a 10 años(30.7%), 3 en la adolescencia, esdecir de 10 a 20 años (7.69%) y 21 en la edad adulta (53.84%). En el 61.53 % el diagnóstico se hizo por encima de los10 años de edad.
De los pacientes diagnosticados como adultos, el 81% eran de 20 a 40 añosy el 19%, de los 46 a los 65 años.
Lugar de nacimiento y procedencia: El dato sobre el lugar de nacimiento estaba documentado en 44 expedientes, y el lugar de residencia en 45.
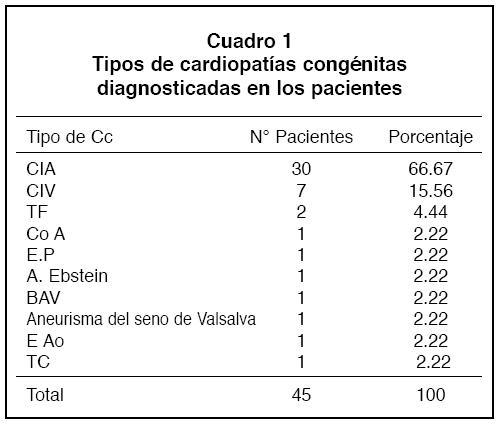
La distribución según el sitio de nacimiento por provincias, fue la siguiente: 16 (36.36%) pacientes habían nacido en San José, 7 (16%) en Puntarenas, 5 (11.3) en Cartago, 4 (9%) en Alajuela, 4 (9%) en Limón, 2 (4.5%) en Heredia, y 2 (4.5%) en Guanacaste, 2 (4.5%) personasnacieron en Nicaragua, 1 (2.2%) en Venezuela y 1 (2.2) en losEEUU (Figura 1).
Figura 1
Porcentaje de pacientes según el lugar de nacimiento
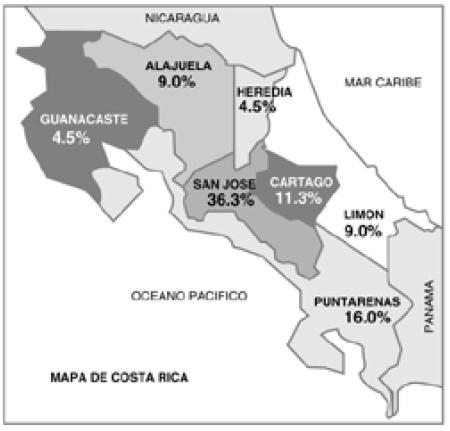
La distribución de acuerdo con el lugar de residencia fue así: 21 pacientes provenían de San José (47%), 5 de Cartago (11%), 5 de Alajuela (11%), 5 de Puntarenas(11%), 4 de Limón (9%), 3 de Guanacaste (7%) y 2 de Heredia (4%). Esta información expresada en tasas por 100000 habitantes de acuerdo a cada provincia, se muestra en la Figura 2. La tasa más alta fue la de San José con un 1.53/100000, seguida por Puntarenas: 1.39, luego Cartago 1.20, Limón 1.17, Guanacaste 1.10, Alajuela 0.78, y finalmente Heredia con 0.59.
Figura 2
Lugar de procedencia por cada 100,000 habitantes
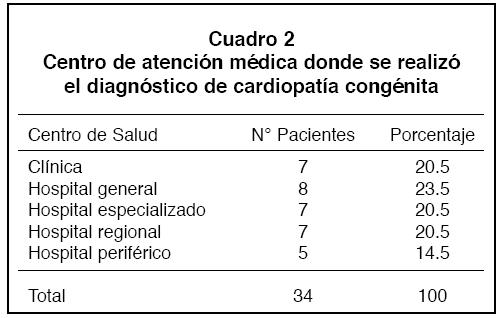
Centro donde se realizó el diagnóstico: Este hecho se documentó sólo en 34 pacientes(75.5%), en 7 de ellosse hizo en una clínica de referencia (20.5%), 8 en hospitales generales(23.5%), 7 en un hospital especializado (HNN; 20.5%), otros 7, en hospitales regionales (20.5%) y los 5 restantes en hospitales periféricos(14.5%).
Estos datos evidencian que el 76.5% de los pacientes llegaron a los hospitales generales con el diagnóstico definitivo establecido (Cuadro 2).
Métodos diagnósticos: En los 45 pacientesse había sospechado la malformación cardiaca por datos clínicos y luego en 42 se realizaron exámenescomplementariosque confirmaron el diagnóstico y definieron el tipo de cardiopatía. El examen definitivo másfrecuente fue el ecocardiograma, que fue realizado en 35 pacientes(77.7%) siendo en su mayoría transtorácico, aunque a 12 pacientes se le realizó también ecocardiograma transesofágico (ETE). Once pacientes fueron cateterizados, siendo este el único examen en tresde ellos.
Defectos asociados: Sólo un paciente tenía un defecto asociado documentado, se trató de un hombre con CIA y Síndrome de Down. Dospacientescon CIA tuvieron severosproblemasde aprendizaje y retraso en el desarrollo psicomotor, pero en losexpedientesno se anotó ningún síndrome genético específico.
Antecedentes familiares de CC: Con respecto a la existencia de familiares con algún tipo de CC, sólo en 8 expedientesse anotó esta relación, sin embargo en la mayoría de loscasosno se especificó el tipo de cardiopatía y si era congénita o no. Sólo una paciente con CIA tiene una prima a quien se le había corregido una TF en el HNN, y que entrevistamos a raíz de esta investigación.
Presentación clínica: Los principales síntomas descritos antes de la cirugía en los 32 intervenidosfueron: disnea de pequeños esfuerzos en 14 pacientes(51%), disnea de grandes esfuerzos en 13 (41%), astenia y adinamia en 10 (31%), palpitaciones en 7 (22%), disnea paroxística nocturna y ortopnea en 6 (19%), edemas en 6 (19%), mareos en 5 (16%), angor en 5 (16%), cianosis en 4 (12%), lipotimias en 3 (9%), cefalea en 1, sudoración en 1 y 4 (12%) enfermos estaban asintomáticos.
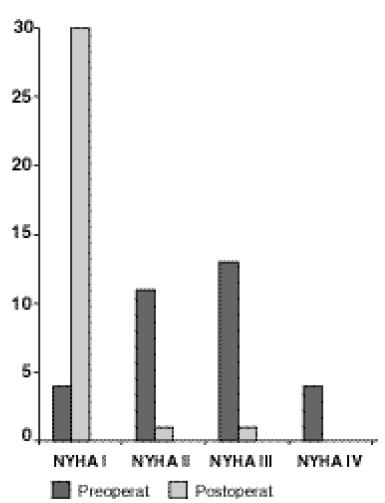
Después de la cirugía, los pacientes asintomáticos eran 27, sin embargo en algunos persistieron los siguientes síntomas: 4 tenían disnea de grandesesfuerzos, 1 con mareos, 3 con astenia y adinamia, 2 tenían angor y 2 pacientespresentaron cefalea. La clase funcional (NYHA), se documentó preoperatoriamente en 32 pacientesintervenidosen 1998. Las clases funcionales predominantes en el preoperatorio fueron la II y la III (24 de 32 enfermos) y la predominante en lospostoperadosfue la I (30 de 32), solamente 2 pacientesno cambiaron de clase funcional (1 de clase I y otro de clase II). En la Figura 3 se puede ver la clase funcional antesy despuésde la cirugía.
Figura 3
Distribución de los pacientes según la clase funcional pre y post operatoria
Paridad e historia obstétrica: De las 34 mujeres, 14 no han tenido hijos, y 12 sí, en 8 casos no se consignó la historia obstétrica y no fue posible averiguarla.
Once tuvieron hijos normales y una tuvo un hijo con CIA. Ocho tuvieron partos por cesárea y 5 tuvieron abortos. Las razones para las cesáreas no estaban relacionadas con la CC de fondo, excepto en una mujer con Tetralogia de Fallot (TF), por lo que se indicó que el parto fuera por cesárea. A una paciente con un Tronco Común (TC) se le recomendó no tener hijos, sin embargo tuvo tres partos vaginales y no se documentó ninguna complicación.
Las 12 mujeres con hijos tuvieron 48 partos vaginales, 8 cesáreas y 5 abortos. Los partosy cesáreas ocurrieron sin complicaciones. Las causasde los abortos no estaban anotadas en los expedientes médicos.
En el caso de los hombres no hay documentación clínica de su paternidad.
Tratamiento: De los 45 pacientes atendidos, 32 fueron exitosamente sometidos a cirugía correctiva en 1998. De los 13 restantes, 4 no eran candidatos a tratamiento quirúrgico según el médico tratante o estaban todavía completando sus estudios, 2 rehusaron someterse a cirugía, 3 mujeres estaban en el posparto inmediato, uno estaba en espera de cirugía y 3 habían sido ya previamente intervenidos.
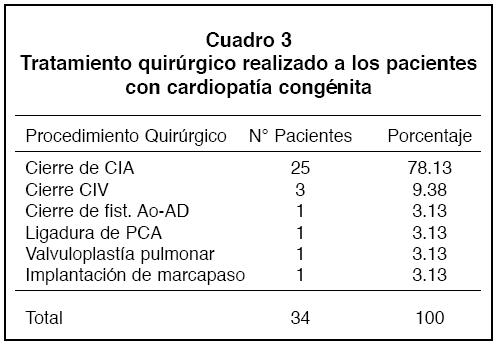
Las intervenciones consistieron en cierre de CIA en 25 pacientes, primariamente en 22 y en 3 con parche de duramadre. Se realizaron también 3 cierres de CIV, y una de cada una de las siguientes intervenciones: cierre de fístula aorto–atrio derecho, ligadura de PCA, valvuloplastía pulmonar e implantación de marcapaso. Después de la cirugía, la gran mayoría de losindividuosse volvieron asintomáticos, o sea de clase funcional I (NYHA). Al aplicar la fórmula de Mcnemar se obtuvo un 95% de certeza en la mejoría de la clase funcional de lospacientessometidosa tratamiento quirúrgico en el año 1998 (Figura 3).
Empleo y capacidad de trabajo: Veinticuatro individuos desempeñan un trabajo normal, y 9 han presentado problemas debidos a su CC, sin embargo solo una paciente tiene incapacidad permanente debido a la severidad de su enfermedad. Se trata de una mujer de 42 años con un TC, cuya CC fue diagnosticada a los 5 años en el HNN, pero que siempre rechazó el tratamiento quirúrgico.
Tres pacientes no trabajan (uno tiene síndrome de Down) y dos son ya pensionados). En 9 pacientes se desconoce este dato.
Discusión
El campo de lasCC esmuy complejo y variado, como ilustración la Oficina de Bioestadística del HNN, en 1998 reportó 32 códigosdiferentesde enfermedadescardíacascongénitasatendidasen suspacientesdurante ese año mismo.13
Clásicamente son los cardiólogos pediatras los encargados del manejo de todos estos enfermos, sin embargo al ir creciendo ellos, operados o no, y al aparecer otros adultoscon CC no diagnosticadas antes, se confirma lo que se preconiza en otras latitudes sobre la necesidad de una subespecialidad dentro de la misma cardiología de adultos, sólo en enfermedades congénitas.5
Los datos de incidencia y prevalencia de CC, disponibles de los organismos nacionales son fragmentados. Aquí las dependencias encargadas de recolectar información (Departamento de Estadística del Ministerio de Salud, Dirección Técnica de Servicios de Salud de la Caja Costarricense de Seguro Social), sólo censan personas egresadas de los hospitales, sin tomar en cuenta la población en general.14,15
Por ejemplo esas dependencias reportan que entre los años1996 y 1999 murieron 32 personas en todos los hospitales nacionales por CC.14,15 Pero la Memoria Anual de 1999 del Ministerio de Salud, que sí incluye losdatosde mortalidad dentro y fuera de los hospitales, reporta, sólo en niños menores de un año de edad, 94 defunciones para 1998.2
Castro del HNN, en 1979 reportó que la incidencia de CC esde 3 a 7 por cada 1000 nacidos vivos,16 aunque según Taussig, en general en el mundo, anda por ahí de 5 a 8 por cada 1000.17 En ese mismo estudio Castro también describió las CC que más frecuentemente se vieron en el HNN en los años previos a su reporte.16 Una publicación reciente del HM, reporta los hallazgos cardiacos en autopsias de óbitos y recién nacidos(RNs).18
De todas maneras la incidencia, prevalencia y variedad de CC en la población nacional es desconocida y a la vez difícil de definir, y más aún aquella que se refiere a los adultos.
Se puede asumir que de cada mil nacidos vivos al menos 5 tendrán CC, o sea que de los 83000 niños nacidos en Costa Rica en 1998, habrá 415 con CC.
Los RNs portadores obvios de CC, son enviados directamente al HNN, poco despuésde nacer, pero habrá muchos sobrevivientes que van a manifestar su enfermedad a lo largo de meses o años, unos antes que otros, y posiblemente ya no en edades pediátricas, complicando la determinación de los datos de incidencia para cada año.
En este trabajo se tomó la muestra de los individuosque consultaron en los hospitales generales durante un año escogido, pero que habían nacido en diferentes años, es decir en cohortes de nacimiento diferentes.
Aunque los 45 pacientesno fueron desglosados por año de nacimiento, a grosso modo pareciera que son pocos los que consultaron. Esto puede sugerir o que hay un buen número de adultos con CC aún sin consultar o que una mayoría de ellos ya habían sido tratados en el HNN.
Este grupo representa una muestra significativa, puesto que loscentrosinvestigadoscubren el territorio nacional y el tipo de patología de estos pacientes solo puede finalmente ser resuelto en ellos.
Características epidemiológicas:
El hallazgo de la preponderancia femenina en este grupo es interesante, ya que otros reportes demuestran una incidencia más alta de CC en varones, y aun más si la atención se centra en ciertas anomalías.
Por ejemplo González y cols., estudiando óbitosy RNsdel HM con CC, reportaron una incidencia del 62% en el género masculino y 32% en el femenino.18 Un estudio inglésreciente, en menoresde un año con CC, reportó 52% en hombresy 48% en mujeres19 y al avanzar en edad losporcentajesse van equiparando.Lasrazonesposiblespara explicar esta diferencia en el sexo de acuerdo a la edad y de la inversión ulterior de loshallazgos, pueden ser: que aunque en nuestro paísnacen máshombresque mujeres, la sobrevida a partir de los50 añosesmayor en mujeresque en hombres, según lo reporta la Memoria Anual de 1999 del MS.3 Por otro lado, esbien sabido que lasmujeresconsultan con mayor frecuencia, y finalmente, la CIA que esla CC másfrecuente en este grupo, también lo esen mujeres, con una relación de 2:1.1, 4
Tipo de CC:
La frecuencia de losdiferentesCC, varía de acuerdo a su distribución mundial y al grupo de edad, y como ya se ha mencionado, la historia natural de cada CC determina si lospacientespueden llegar o no a la vida adulta.
Fuster, en un estudio de CC en adultosrealizado en 1980, encontró que la CIA era la CC más frecuente.19
Suresh et al., revisaron 6 series asiáticas y en 5 la CIA fue la primera o la segunda en frecuencia, la CIV lo fue en 3 y la TF en 3.20 El orden de frecuencia hallado en este estudio (CIA,CIV, TF), concuerda con algunas de estas investigaciones.
Edad del diagnóstico: En esta muestra el diagnóstico de la CC se realizó en un 61.53% de los pacientesen edades superiores a los10 años, y en casi la quinta parte de ellos se hizo entre los 46 y los 65 años. En contraposición, Hoffman en una recopilación de 9 estudios de diferentes áreas, reporta una detección del 90 al 100% de las CC durante los primeros 5 años de vida.6 Fenómeno confirmado por Robida de Qatar quien tambien reporta una detección del 100% a los 5 años de edad.18
Los datos obtenidos en esta investigación, señalan por lo tanto un retraso en nuestro medio, en cuanto a la edad del diagnóstico de las CC.
Si vemos ahora el tipo de CC, tenemos que en todas en las que el diagnóstico se hizo tardíamente, esta era una CIA.
Aunque estos individuos tienen un soplo sistólico de por vida, y otros datos semiológicos típicos, ellos pueden pasar desapercibidos al explorador no entrenado, fenómeno ya reconocido en la literatura.36 El adulto joven con una CIA no corregida y presión arterial pulmonar normal suele ser asintomático. Aunque los pacientes con CIA no corregida, generalmente sobreviven hasta la edad adulta, su expectativa de vida no es normal, sólo cerca de 50% sobrevive más allá de los 40 años. La mortalidad después de los 40 años es de casi 6% por año.3
Procedencia: La razón para explicar porque San José tuvo las tasas más altas de procedencia y de nacimiento no se conoce. Podría especularse que sus habitantes tienen mejor acceso a los centros de salud y consultan más, su población es más mezclada y los pacientes a menudo cambian su dirección para no ser referidosa otros centros, por lo que algunos podrían no ser de esta provincia.
Centro donde se realizó el diagnóstico: El 76.47% de los pacientes llegaron a los hospitales generales con el diagnóstico hecho. Esto evidencia que a pesar de que existen centros con menor tecnología y menos profesionales entrenados en este campo, en comparación con los hospitales generales o especializados (HNN), el diagnóstico pudo realizarse en forma acertada, hecho que permitió referir a estos pacientes para su posterior manejo y tratamiento a los centros respectivos, ahorrando tiempo y dinero para los pacientesy los distintos centrosde salud.
Defectos asociados: Se reporta que el Síndrome de Down ocurre en 1 de cada 660 nacidos vivos, frecuentemente son productos de madres añosas y que de 40 a 50% para unos y de 25 a 60% para otros, estos niños nacen con defectos cardiacos, sobre todo del tipo CIA ostiumprimum.6 En nuestra serie hubo un paciente con Down y CIA, cuya madre lo tuvo a los 41 añosde edad.
Método diagnóstico:
El diagnóstico de las CC se hizo en primer lugar con datos clínicos. El hallazgo de distintos síntomas y signos físicos permite al clínico tener una aproximación diagnóstica, sin embargo, existen variaciones con respecto a la formación y experiencia del cuerpo médico, hecho que afecta e interfiere en la detección de las distintas CC.
Otros aspectos que intervienen son, la calidad de servicios de salud, el método diagnóstico utilizado y el tipo de CC, ya que algunas se presentan con un cuadro florido desde el nacimiento, mientras que otras pueden ser asintomáticas y no ser detectadas en un examen físico común.6
En el estudio de las CC los exámenes de rutina y tamizaje, más usados son la radiología de tóraxy el EKG, pero los definitivos son el ECO, el cateterismo cardíaco y la cardioangiografía.3,22,23 Estos fueron los mismos utilizados por los médicosque estudiaron esta serie de enfermos, quedando la ETE y el cateterismo para aquellos pacientes en los cuales había dudas o en los que otros estudios no habían podido aclarar con exactitud la anatomía.
Harrison en un estudio sobre los cambios ocurridos en los adultos con CC refiere que el diagnóstico se hace con la radiografía de tórax y con el ECO, y en caso de duda, se utiliza el ETE y el cateterismo cardiaco como segunda opción.24
Paridad: Existen CC cianógenasy acianógenas que permiten que el individuo llegue a la vida adulta y a un ser capaz de procrear,24 como sucedió en 12 de nuestras pacientes.
A pesar de que las pacientes que tuvieron hijos, no reportaron complicaciones obstétricas, la presencia de una CC puede representar problemas especiales para la mujer embarazada. Existen riesgos de muerte durante el parto o en el posparto inmediato y también cuando se realiza una cesárea, aunque esta cirugía debería producir menos estrés que el parto mismo y el período expulsivo vaginal.3,23
A consecuencia de los riesgos del embarazo, en otros países algunos médicos recomiendan el aborto precoz en ciertas mujeres con CC, por lo cual ellas deben recibir asesoramiento sobre control de natalidad.1
El consejo prenatal de la paciente debe incluir información sobre riesgos de recurrencia para los descendientes.1
Se ha demostrado en algunos estudios que los varones se preocupan tanto como las mujeres por el riesgo de tener hijos con una CC. En especial, los varones pueden experimentar incertidumbre sobre su capacidad para convertirse en el sostén de su familia, dados los desafíos que afrontan para obtener un empleo fijo o seguro de salud y de vida como ocurre en algunos países.1
El estado de salud de los hijos de las pacientes aquí estudiadas, era normal, excepto una mujer cuyo hijo heredó una CIA. En diversos estudios se ha demostrado un incremento en el riesgo del feto de una paciente con CC. En general el riesgo de recurrencia de CC en los hijos de padres afectados fluctúa de 2 a 4%.25
Se ha documentado que cuando una persona tiene una CIA, el 2.6% de sus niños tenía el mismo defecto, una incidencia que es 37 veces superior a la esperada. Para la CIV, el riesgo es 21 veces mayor que en la población general.25 Otros estudios sugieren que los defectos que afectan el flujo cardiaco izquierdo (Co/A, E/Ao y la Hipoplasia del VI ) son más frecuentes entre familiaresque otros tipos de defectos.17 Castro cita que los datos del cuestionario del Natural History Study II, indican que la tasa de recurrencia de CC en hijos de personas con E/Ao era de 2.2%. Similar al de las madres con CIV 2.9% y EP con 2.89% de riesgo.
Debido a lo anteriormente expuesto, los hijos de pacientes con CC debieran ser referidos a los centros de salud urbanos para una valoración especializada, aún ante la ausencia de síntomas.
Tratamiento: La mejoría de lossíntomas después de la intervención demuestra la bondad del tratamiento quirúrgico, el cual tiene un mayor beneficio si se realiza a edades tempranas.3,5,6,24
Empleo y capacidad de trabajo:
A pesar de que sólo 9 de los pacientes estudiados tienen problemas para desempeñar su trabajo. Las personas que sufren de una CC, tienen múltiples limitaciones. En Estados Unidos por ejemplo, un tercio de los adultos con CC, tienen problemaspara conseguir segurosde vida, aún cuando éstos sean capaces de trabajar.1,5
En nuestro medio, esta situación no se observa, graciasa que tenemosun sistema de seguridad social, que cubre a todos los individuos sin importar condicionesde salud preexistentes.
Las oportunidades de trabajo para este tipo de personas se ven influenciadas por la educación que tengan, ya que algunas CC pueden afectar el intelecto, también es importante el trabajo a realizar, si el paciente ha sido o no operado y la disposición del empleador, ya que muchas veces puede haber discriminación hacia estas personas.1
También es necesario recordar el aspecto psicológico. Los pacientes con CC sufren por su condición, por los tratamientos a los que deben ser sometidos, y por el miedo a trasmitir a su progenie su cardiopatía, experimentando sentimientosde inseguridad y vulnerabilidad con respecto a su futuro.1,5
En conclusión, en esta muestra de pacientes egresados de los hospitales generales con CC durante 1998, predominó el sexo femenino, la CIA fue la CC más frecuente y el diagnóstico en la mayoría de los enfermosse realizó en la edad adulta. Finalmente debido a la incidencia mayor de CC, en la descendencia de los pacientes con CC, estos individuos deben ser aconsejados tempranamente sobres estos riesgos y sus hijos deben ser examinados mas detenidamente para descubrir la cardiopatía si es que la tienen.
Abstract
The incidence and prevalence of congenital heart disease (CHD) in Costa Rica and other underdeveloped countries is unknown. In spite of having a broad nationwide heath coverage we still see untreated adults with CHD. Given the demographic growth and the decline in infancy mortality the modern physician faces a larger number of people with CHD, whom he has to diagnose, refer or advise. In order to learn more about our own reality, particularly of adults with CHD, we studied all the patients discharged fromthe metropolitan area hospitals. We choose the year of 1998 in order to have recent information, and to provide us with al least 2 years of follow up data.
There were 45 adults, 75% were women and 25% were males, with a range of 15 to 64 years of age. Most of them had been born and came from the province of San Jose.
Sixty six percent of the patients had atrial septal defect, 11.5% had ventricular septal defect, 4.4% had Tetralogy of Fallot and there wasa case each of aortic coarctation, aortic stenosis, Ebsteins anomaly, truncusarteriosus, congenital AV block, and sinus of Valsalva fistula.
In 61,53% the diagnosis was made when they were older than 10 years of age. In 81% of the adults the diagnosis was made when they were 2040 years old and in 19%, when they were between 46 to 65 years old.
Echocardiography was the most utilized and definitive exam.
Of the 34 women, 12 had children, there were 48 vaginal deliveries and 8 csections without mortality or complications. One ASD patient had a child with ASD. Thirty two patients were corrected during the year of the study and all survived their surgeries. Obstetrical indications and medical treatment were the other reason for hospital admission. The NYHA functional class was I in 30 of 32 individual submitted to surgery.
Twenty four subjects were employed and 2 had already retired. One patient with Downsand one with truncusarteriosus who had refused surgery were disabled. Nine individuals had problems performing a job and in 9 this information was not recorded.
From these data we can gather that the diagnosisof CHD is been made late in comparison with other countries. Most of the patients are referred to the metropolitan area with the diagnosis already made. The surgical treatment is highly successful and the patients are able to join the work force and procreate normally.
Referencias
1. Skorton DJ, Garson A Jr. Clínicas Cardiológicasde Norteamérica. En: Cardiopatías Congénitasen Adolescentesy Adultos. 1ed. México D.F: Mc GrawHill Interamericana; 1993; 587755. [ Links ]
2. Pardo R, Carvajal X. Memoria Anual 1999. Ministerio de Salud. Costa Rica. Departamento de Publicacionese Impresiones MS 2000;1: 119. [ Links ]
3. Crawford MH. Diagnóstico y Tratamiento en Cardiología. En: Elyse Foster, Cardiopatías congénitas en adultos. México D.F: Manual Moderno S.A de CV; 1997; 419468. [ Links ]
4. Murray G, Baron MD. Plain Film diagnosis of common cardiac anomaliesin the adult. Radilogic Clinics of North America. W.B. SaundersCompany 1999; 37:120. [ Links ]
5. Perloff JK. Congenital heart disease in adults. Circulation 1991; 84:1881-1890. [ Links ]
6. Hoffman JIE. Incidence of congenital heart disease: Postnatal incidence. Pediatric Cardiol 1995;16(3): 1031-13. [ Links ]
7. Jaramillo J. Principiosde Gerencia y Administración de ServiciosMédicos. 1a edición. Editorial Universidad de Costa Rica. Costa Rica1998; 8993. [ Links ]
8. Instructivo para presentar propuestasde investigación CCSS. Unidad de investigación científica, Hospital México. CCSS 1997. [ Links ]
9. Clasificación Estadística Internacional de lasEnfermedadesy ProblemasRelacionadoscon la Salud. 10ª. Revisión. Organización Panamericana de la Salud. Oficina Sanitaria Panamericana. Oficina de la Organización Mundial de la Salud 1995; 7: 700710. [ Links ]
10. The Criteria Committee of the New York Heart Association. Functional capacity and objective assessment. En Nomenclature and criteria for diagnosisof diseasesof the heart and great vessels. 9a. Ed. Boston. Little Brown & Company 1994; 254. [ Links ]
11. Friedman WF, Child J. Congenital Heart Disease in the Adult, en HarrisonsPrinciplesof Internal Medicine. 14ava. Ed. Fauci, Braunwald, Isselbacher, Wilson (Editores.). Philadelphia, Saunders1998; 13001307. [ Links ]
12. Gomez M. Elementosde Estadística Descriptiva. 3ra. Edición. Costa Rica. Editorial UNED 1998; 9497. [ Links ]
13. Boletín de la Oficina de Biestadística Hospital Nacional de Niños. San José, Costa Rica, 1998. [ Links ]
14. Dirección Técnica de Serviciosde Salud CCSS. Sistema de egresoshospitalarios. Sección de información biomédica. Costa Rica1996; SEGI07OD. [ Links ]
15. Boletín Departamento de Estadística. Unidad Sensorial de Planificación. Ministerio de Salud. 19961999. [ Links ]
16. Castro A. CardiopatíasCongénitas. Revista Médica Hospital Nacional de Niños1979 (Edición extraordinaria): 181194. [ Links ]
17. Taussig H. Congenital Heart Disease after surgery. En: Engle Allen MD, J. Perloff. World survey of the common cardiac malformations: developmental error or genetic variant?. 1sted. New York: York Medical Books1983; 142. [ Links ]
18. González L, Salazar L, Salazar C. CardiopatíasCongénitasen el Hospital México. Revista Médica de Costa Rica y Centroamérica 2000; 551: 4756. [ Links ]
19. Fuster V, Brandenburg RO, McGoon DC, Giuliani ER. Clinical approach and management of congenital heart disease in the adolescent and adult. Cardiovasc Clin 1980; 10: 161197. [ Links ]
20. Suresh V, Rao AS, Yavagal ST. Frequency of variouscongenital heart diseases: analysisof 3790 consecutively catheterized patients. Indian Heart J 1995; 47: 125128. [ Links ]
21. Robida A, Folger GM, Hajar HA. Incidence of congenital heart disease in Qatari children. Int J Cardiol 1997; 60: 1921. [ Links ]
22. Massie BM, Amidon TA. Capítulo 10 Corazón. En: Tierney LM, McPhee SJ (Eds). Diagnóstico y tratamiento. 37ava Edición México. Manual Moderno 1999; 332. [ Links ]
23. MossAJ. Cluesin diagnosing Congenital heart disease. West J Med 1992; 156: 392398. [ Links ]
24. Harrison WL. Changesposed by the adultswith Congenital Heart disease. Adult Intern Med 1996; 41: 43795. [ Links ]
25. Burn J, Brennan P, Little J, Holloway S, Coffey R, Somerville J, et al. Recurrence risksin offspring adultswith major heart defects: resultsfromfirst cohort of British collaborative study. Lancet 1998; 351: 311-316. [ Links ]Abreviaturas: AV, atrio–ventricular; CC, cardiopatía (s) congénita (s);

