










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Costarricense
versión On-line ISSN 0001-6002versión impresa ISSN 0001-6012
Acta méd. costarric vol.42 no.3 San José sep. 2000
Resumen
La Linfangioleiomiomatosis pulmonar es una enfermedad muy rara, que afecta sólo mujeres en edad reproductiva. Se presenta con disnea progresiva, pneumotórax a repetición y, ocasionalmente, con hemoptisis y quilotórax. El TAC de alta resolución muestra quistes pulmonares bilaterales de tamaño variable hasta bulas francas. Histológicamente, se aprecian múltiples cavidades de paredes finas, dilatación de vasos linfáticos, venas, arterias, bronquiolos y sacos alveolares, por proliferación de haces de músculo liso que comprime las estructuras antes mencionadas y que causa entonces su dilatación.
El pronóstico es malo, ya que las pacientes desarrollan insuficiencia respiratoria, lo que las conduce a la muerte. La enfermedad se asocia a la ingesta de estrógenos, píldoras anticonceptivas y se exacerba con el embarazo-, por ello se ha tratado con medróxi-progesterona y tamoxifén, con estabilización de la evolución en algunos pero no en todos los casos. Eventualmente, algunas pacientes pueden necesitar transplante pulmonar. Presentamos el caso de una mujer de 38 años con tres episodios de pneumotórax espontáneo y documentación radiológica e histológica de linfangioleiomiomatosis pulmonar.
Descriptores
Pneumotórax, disnea, linfangioleiomiomatosis.
Esta rara enfermedad fue descrita primero en la literatura anatomo-patológica en 1937 como "cirrosis muscular pulmonar" y luego en la radiológica en 1942, a raíz de casos de mujeres con insuficiencia respiratoria terminal, cambios quísticos pulmonares y extensa proliferación de tejido muscular liso en los mismos.1 Hoy en día se le conoce como linfangioleiomiomatosis pulmonar (LLMP).
La entidad afecta solamente mujeres y se presenta corrientemente con disnea progresiva, episodios de pneumotórax espontáneo, y algunas veces con hemoptisis y quilotórax.
Como ocurre en mujeres de edad fértil, y sus exacerbaciones se han relacionado con el embarazo, la ingesta de estrógenos y las píldoras anticonceptivas, se ha sugerido que las hormonas sexuales ocupan un papel importante en su aparición.1-2
El músculo liso anormal crece alrededor de los conductos aéreos, de los vasos sanguíneos y de los linfáticos, y esta distribución explica las manifestaciones clínicas de la enfermedad. La compresión de las vías aéreas causa obstrucción al flujo del aire y eventualmente hay destrucción de las paredes alveolares, con la formación de espacios quísticos y bulas, que al romperse producen pneumotórax; al ser rodeados los vasos pulmonares, se produce congestión y hemoptisis, y la obstrucción linfática conduce a la producción de quilotórax.
Esta enfermedad se puede presentar en asociación con otras condiciones, por ejemplo el complejo de esclerosis tuberosa o los angiolipomas renales.2-5 Reportamos aquí, el primer caso documentado en nuestro país, de esta entidad nosológica.
Caso clínico
Paciente femenina, de 39 años, ama de casa, blanca, no furiiadora, con antecedentes de tres episodios de pneumotórax espontáneo, dos derechos (junio y setiembre de 1997) y 1 izquierdo (marzo de 2000), todos asociados a la menstruación, tratados y resueltos con toracostomía, excepto por el penúltimo que requirió además pleurodesis química. A pesar de que el colapso pulmonar se había resuelto, se presentó a la Consulta Externa quejándose de disnea, ahora de pequeños esfuerzos y de reposo y también de dolor en el hemitórax derecho. Por esta razón se practicaron nuevas radiografías de tórax y se planteó la posibilidad de pneumotórax catamenial.6
Las placas convencionales no mostraron patología obvia. Se practicó también un ultrasonido de abdomen y de pelvis para investigar la posibilidad de endometriosis, pero este estudio fue negativo. La tomografía axial computarizada mostró múltiples quistes pequeños de distribución difusa bilateral y una lesión mayor en el campo pulmonar derecho, francamente bulosa. Los septos interlobulares se apreciaron discretamente engrosados pero el parénquima pulmonar entre los quistes estaba respetado (Figura 1). En otras áreas había quistes de paredes delgadas sin engrosamiento (Figura 2). No había pneumotórax ni quilotórax. Los hallazgos radiológicos sugirieron fuertemente el diagnóstico de LLMP.
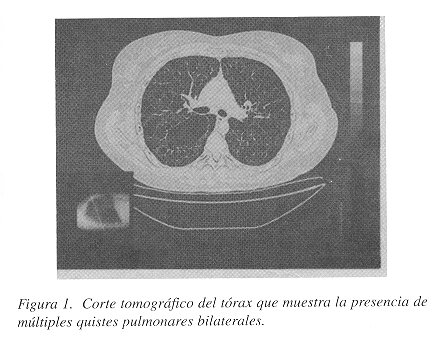
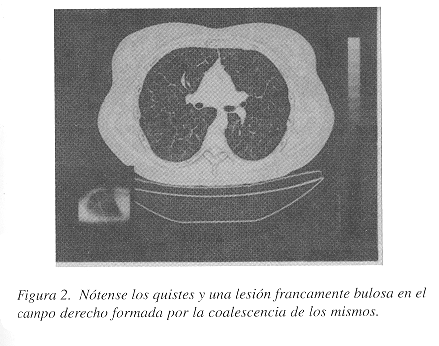
Las pruebas de espirometría mostraron un patrón mixto, siendo el componente obstructivo de grado severo. Debido a lo poco frecuente de esta entidad y para estar seguros del diagnóstico, se recomendó una biopsia pulmonar. Esta se obtuvo mediante una minitoracotomía anterior izquierda. No había derrame pleural, y la língula y el resto del pulmón que podía visualizarse, mostraban múltiples lesiones subpleurales bulosas no mayores de 4 mm. de diámetro, observables también en el parénquima pulmonar al corte.
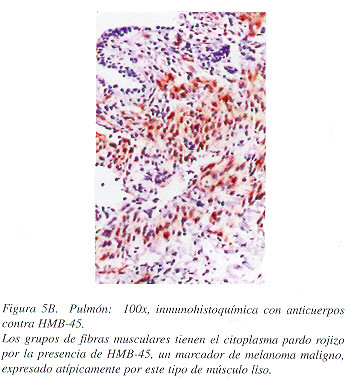
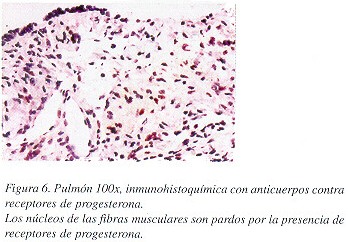
Macroscópicamente, la cuña de tejido pulmonar midió 2 x 1.5 x 1 cm. y presentaba un aspecto esponjoso, debido a la presencia de cavidades vacías de hasta 0.4 cm. de diámetro, de paredes finas y enfisema centrolobulillar. Microscópicamente se encontraron múltiples vasos linfáticos, venas, arterias, bronquiolos terminales y sacos alveolares dilatados, con pequeñas zonas nodulares de proliferación de haces de fibras de músculo liso en sus paredes (Figuras 3 y 4). No se observó contenido en las cavidades. Las fibras de músculo liso eran de aspecto maduro y reaccionaron con el marcador de actina para músculo liso (Figura 5A), con el de melanoma maligno HMB-45 (Figura 5B) y con el de receptor de progesterona (Figura 6), y no expresaban ni proteína S-100; ni receptores de estrógenos.
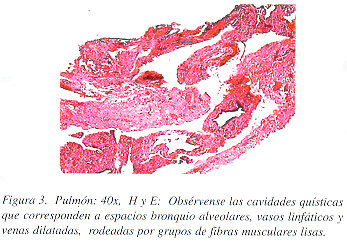
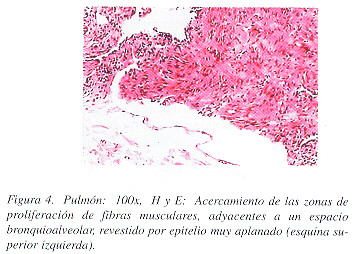

Los hallazgos anatomo-patológicos confirmaron el diagnóstico de LLMP. La paciente fue informada de su enfermedad y se le propuso un plan terapéutico como se recomienda en la literatura.7 Sin embargo, nos confió que había también consultado a un médico naturalista y que ya había iniciado un tratamiento recomendado por él.
Discusión
La LLMP es una enfermedad muy rara de reporte casi anecdótico. De hecho, para entender mejor esta entidad la Clínica Mayo y la Universidad de Stanford, ambos grandes centros de referencia, establecieron un re-istro de casos, recogiendo de 1976 a 1990 solamente 32 pacientes debidamente estudiados y todos seguidos por ellos mismos.1 El Grupo Cooperativo Francés que comprende unos 200 pneumólogos repartidos en todo el territorio de ese país, recogió información de 1995 a 1997, y reportó 69 pacientes ya conocidas o nuevas, vivas o muertas, que en algún momento habían sido vistas por ellos. En nuestro medio esta es la primera vez, hasta donde hemos podido investigar, en que se documenta y se reporta esta entidad.
Se distingue en los estudios mencionados un aumento de la frecuencia de la enfermedad de los 30 a los 34 años de edad. Se ha diagnosticado más tardíamente, pero queda la duda del verdadero inicio del cuadro, ya que el diagnóstico está en función de su sintomatología.
El estudio radiológico en estos casos, se inicia con la radiografía de tórax, la cual puede mostrar aumento del volumen pulmonar, hiperinsuflación y un patrón intersticial y reticular fino, que en estadios avanzados es generalizado; estos hallazgos son poco específicos.2, 8 En 3% de las pacientes la radiografía de tórax es normal.2 La tomografía axial computarizada (TAC) y la tomografía de alta resolución exhiben mejor que la radiología convencional los hallazgos de esta enfermedad. En etapas tempranas el TAC muestra la presencia de quistes, distribuidos difusamente en ambos pulmones.9 Estos van desde pocos milímetros de diámetro hasta varios centímetros; cuando la enfermedad avanza, los quistes coalescen, formando lesiones más grandes de contornos lobulados,10 como se apreció en nuestro caso. Se ha reportado también la apariencia de vidrio esmerilado hasta en un 52% de los pacientes, puede haber pneumotórax, y derrame pleural.
Los quistes grandes de paredes ondulantes que se ven en algunos casos, son similares a los vistos en el granuloma
eosinófilo.2,9
El diagnóstico histopatológico de la LLMP es difícil, ya que las zonas de dilatación de estructuras vasculares y bronquioalveolares, junto con las áreas de proliferación de fibras musculares lisas dan un aspecto hamartomatoso a esta lesión. Si la biopsia ha sido obtenida transbronquialmente, puede confundirse con una fibrosis intersticial.
Hay tres elementos que pueden ayudar a entender esta entidad: a) el análisis inmunohistoquímico, el cual establece la identidad de las fibras musculares, pero muestra una expresión atípica de un marcador para melanocitos neopiásicos y una expresión de receptores hormonales; b) la asociación con angiomiolipoma renal uni o bilateral en un 8 a un 46% de las pacientes,2-3 con esclerosis tuberosa y por último c) la correlación con la estimulación hormonal.
En los análisis inmunohistoquímicos, la expresión del marcador de actina para músculo liso por las celulas fusiformes presentes en las zonas nodulares en la biopsia pulmonar de esta paciente, y la falta de expresion de la proteína S- 1 00 (un marcador para fibras nerviosas e histiocitos), apunta que las células son de origen muscular liso y no de origen nervioso (neurofibromatosis) o histiocíticas (histiocitosis X). El marcador CD1, que es más específico para las células de Langherhans (histiocitos de la histiocitosis X), no está disponible en nuestro medio. Sin embargo, no se encontraron otras características histológicas para el diagnóstico de esta última entidad, por lo que se descartó. La expresión de HMB-45, un marcador de melanocitos, es inesperada en fibras musculares lisas, pero este inmunofenotipo también se ha descrito en las fibras musculares del angiolipoma renal.11 Aun más, este hallazgo ayuda a establecer el diagnóstico en muestras pequeñas transbronquiales y a descartar otras posibilidades como el enfisema panacinar, la histiocitosis X o la fibrosis pulmonar intersticial idiopática, condiciones en las cuales los fibroblastos no expresan este marcador.
La esclerosis tuberosa es un complejo sindrómico de transmisión autosómica dominante, en el que 1-2% de los casos existen lesiones pulmonares, las cuales son idénticas a las de LLMP. 1,2 Estas aparecen casi exclusivamente en mujeres en la tercera y cuarta década de la vida. Tanto en esclerosis tuberosa como en la LLMP se ha demostrado pérdida de la heterocigocidad en el cromosoma 9 y 16, en la primera y cromosoma 16 en la segunda. Esto sugiere la pérdida de un gen supresor de tumor en ambas condiciones.2 Se requerirá el análisis de la secuencia genética para determinar si éstas comparten el mismo defecto genético.
Como la LLMP se presenta en mujeres en edad reproductiva, y se ha visto mejoría clínica post-ooforectomía, tratamiento con medroxi-progesterona o terapia antihormonal, la influencia estrogénica en esta condición es clara. El análisis de receptores hormonales en los tejidos biopsiados de las pacientes con esta enfermedad los ha demostrado por diversos métodos.12 Actualmente, la inmunohistoquímica con anticuerpos dirigidos contra receptores hormonales es un método más sencillo y accesible en nuestro medio. En nuestra paciente se demostró la presencia de receptores de progesterona en los núcleos de las células musculares lisas, como se ve en la Figura 5.
El tratamiento recomendado actualmente es la administración mensual de 400 mgs. de acetato de medroxi-progesterona intramuscularmente cada mes; algunos autores recomiendan simultáneamente utilizar tamoxifén 1,2,5 y otros en cambio prefieren usar agonistas de la hormona liberadora de gonadotrofina, como la triptolina. Las pacientes deben ser seguidas con pruebas de espirometría seriadas para verificar su evolución y respuesta al tratamiento. No todas las pacientes responden a los métodos mencionados. Para aquellas que se presentan tardíamente o que no responden a la terapia son candidatas a transplante pulmonar.
Abstract
Pulmonary lymphangioleiomyomatosis is an uncommon disease, that affects only women of child-bearing age. The patients present themselves with progressive dyspnea, frequent episodes of spontaneous pneumothorax and, occasionally, with hemoptysis and chylothorax. High resolution CAT sean shows bilateral lung cysts of variable size up to frank bullae. Histologically, numerous thin walied cavities are seen, and dilatation of lymphaties, veins, arteries, bronchioles and alveolar sacs with extensiva smooth muscle proliferation, which engulfs the previously mentioned structures and causes their dilatation.
The patients eventually develop respiratory insufficieney and die. This disease is associated with estrogen intake, contraceptivas and becomes worse during pregnancies. Thus, it has been treated with medroxi-progesterone and tamoxifen, with stabilization of the clinical course in some but not in al] the cases. We present here the case of a 38 year old female with three episodes of spontaneous pneumothorax and radiological and microscopic documentation of pulmonary lymphangioleiomyomatosis.
Referencias
1 . Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis. Clinical course in 32 patients. NEJM 1990; 323 (18) 1254-1260. [ Links ]
2. Urban T, Lazor R, Lacronique J, Murris M, Labrune S, Valeyre D, Cordier JF. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Medicine 1999; 78: 321-327. [ Links ]
3. Bernstein SM, Newell JD, Adamczyk D, Mortenson RL, King TE, Lynch DA. How common are renal, angiomyolipomas in patients with pulmonary lymphangioleiomyomatosis? Am J Respir Crit Care Med 1995; 152:2138-43. [ Links ]
4. Jao J, Gilbert S,Messer R. Lymphangiomyoma and tuberous sclerosis. Cancer 1972; 29: 1188-1192. [ Links ]
5. Castro M, Shepherd CW, Gomez MR, Lie JT, Ryu H. Pulmonary tuberous sclerosis. Chest 1995; 107: 189-95. [ Links ]
6. Salazar C, Araya H, Alvarado EM, Soto L. Pneumotórax catamenial. Rev Med de CR y CA. 1997; LIV(541): 141-144. [ Links ]
7. Zapatero J, González-Palacios F, Ortíz de Saracho J, Sánchez J, Candelas J. Linfangioleiomiomatosis pulmonar con respuesta al trata miento con medroxi-progesterona y tamoxifén. Cirugía Española 1995, 58: 266-268. [ Links ]
8. Moss A. Computed tomography of the body. Philadelphia: W, B Saunders, 1992. [ Links ]
9. Webb WR. Advances in chest radiology. In The Radiologic Clinics ot North America. Philadelphia: W.B. Saunders. 1994: 32:748-749. [ Links ]
10. Burgener F, Kormano M. Diagnóstico por TAC. Madrid: Ed Marban Libros. 1998. [ Links ]
11. Chan JKC,Tsang WYW, Pan MY, Tang MC,Pan SW, Fletcher CDM. Lymphangiomyomatosis and angiomyolipoma. Closely related entities characterized by hamartomatous proliferation of HMB-45 positiva smooth muscle. Histopathology 1993; 22:445-455. [ Links ]
12. Graham ML, Spelsberg TC, Dines DE, Payne WS, Bjornsson J, Lie JT. Pulmonary lymphangiomyomatosis: with particular reference to steroid-receptor assay studies and pathologic correlation. Mayo Clin Proc.1984: 59:3-11. [ Links ]
Abreviaturas
LLMP, Linfangioleiomiomatosis Pulmonar; TAC, Tomografía Axial Computarizada.
1. Servicio de Cirugía de Tórax y Cardiovascular, Hospital México.
2. Servicio de Anatomía Patológica. Hospital México.
3 Servicio de Imágenes Médicas, Hospital México.
Correspondencia: Carlos Salazar Vargas. Apdo. 1826-1250 Escazú.

