









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
La deficiencia congénita de factor VII es uno de los desórdenes congénitos de la coagulación más comunes. Se caracteriza por su herencia autosómica recesiva, con penetrancia variable1,2 y tiene una prevalencia a nivel mundial de 1:300,000- 1:500,000.3 El gen en el cual ocurren las mutaciones es el F7, localizado en el cromosoma 13 (13q34).4,5
Un tercio de los pacientes con deficiencia de factor VII tiende a mantenerse asintomático toda su vida, por lo que la prevalencia puede ser mayor a la reportada.4 La edad promedio de diagnóstico de este defecto hereditario es alrededor de los 8 años y los síntomas varían dependiendo del caso.3 Existen manifestaciones menos graves de este trastorno, las cuales incluyen: epistaxis, gingivorragia, hematomas, metrorragia y hematuria; mientras que las manifestaciones graves del padecimiento incluyen: hematomas extensos, hemartrosis, sangrados gastrointestinales y hemorragias del sistema nervioso central.8 El sangrado grave usualmente ocurre posterior al nacimiento y se presenta a nivel del sistema nervioso central con una frecuencia de 2,5%. Rara vez se reportan los casos de pacientes con sangra- dos cerebrales neonatales que logran sobrevivir.4,6
A continuación, se presenta un caso pediátrico con el que se busca estimular en la comunidad médica la sospecha clínica de deficiencia de factores de coagulación ante cuadros clínicos de sangrados inusuales en edad pediátrica, en particular los cerebrales, así como la conciencia de la importancia de esta sospecha clínica en el abordaje inicial de estos casos poco comunes.
Presentación de caso
Paciente masculino, nacido por cesárea intra- parto, con una edad gestacional de 37 semanas y 5 días, un peso al nacer de 3500 kg, talla de 51 centímetros, circunferencia cefálica de 33,5 centímetros y Apgar 9/9, sin datos de sufrimiento fetal y con esquema de vacunación completo. Posee el antecedente heredofamiliar de muerte de hermano a los 4 días de nacido por hemorragia intracraneal sin etiología identificada y con padres no consanguíneos. A los 14 días de nacido, se le llevó al servicio de emergencias por sangrado umbilical, el cual persistía después del desprendimiento del cordón. La madre consultó en tres ocasiones y se le dieron recomendaciones con manejo ambulatorio; sin embargo, el sangrado en el sitio del cordón umbilical persistió, por lo cual la madre consultó de nuevo a los 21 días de nacido el niño. Se decidió ingresar al paciente y realizar estudios. El abordaje inicial incluyó la toma de tiempos de coagulación, lo que mostró alteración a expensas del tiempo de protrombina (TP), con tiempo de tromboplastina parcial (TTP) normal y fibrinógeno normal (Cuadro). Se decidió administrar vitamina K en 3 ocasiones, pero al permanecer prolongado el TP, se resolvió transfundir plasma fresco congelado (PFC). No obstante, el paciente persistió con sangrado y alteración del TP, lo cual hizo que se sospechara de un defecto hereditario de la coagulación. Se interconsultó a Hematología Pediátrica y se realizaron cuantificaciones de factores de coagulación, con lo que se confirmó la deficiencia congénita severa de factor VII. Se colocó una dosis de rFVIIa, con lo cual aumentó el nivel de factor VII (Cuadro).
Para este momento, el paciente ya no presentaba sangrado, por lo cual se decidió proseguir sin profilaxis hasta que llegara a una edad donde, por su movilidad, existiera riesgo de trauma. A los 33 días de nacido, el paciente presentó una infección por SARS-CoV-2 que ameritó internamiento, pero que se resolvió sin presentar secuelas. A los 2 meses de nacido, el paciente presentó historia de 24 horas de evolución de irritabilidad y rechazo al alimento.
Se le solicitó una tomografía axial computarizada urgente que evidenció hemorragia en sistema nervioso central a nivel frontal derecho, motivo por el cual se le realizó una craneotomía con hemilobectomía frontal derecha. Debido a esta manifestación de sangrado grave, se tomó la decisión de iniciar profilaxis con rFVIIa diaria hasta la resolución del sangrado cerebral. A los 2 meses y 10 días, se le realizaron estudios moleculares basados en secuenciación masiva de nueva generación (NGS por sus siglas en inglés). El análisis determinó dos variantes heterocigotas: F7, intrón 5, c.430+1G>A y F7, intrón 8, c.805+1G>A, definidas en conjunto como patogénicas según los criterios PVS1, PMS2 y PP5 del American College of Medical Genetics. (Figura)7 Actualmente, el paciente continúa con profilaxis 5 días de la semana con rFVIIa 200 µg/ día IV (280 µg/kg) de lunes a viernes a través de catéter subclavio externo permanente. El paciente no ha vuelto a presentar ninguna manifestación de sangrado.
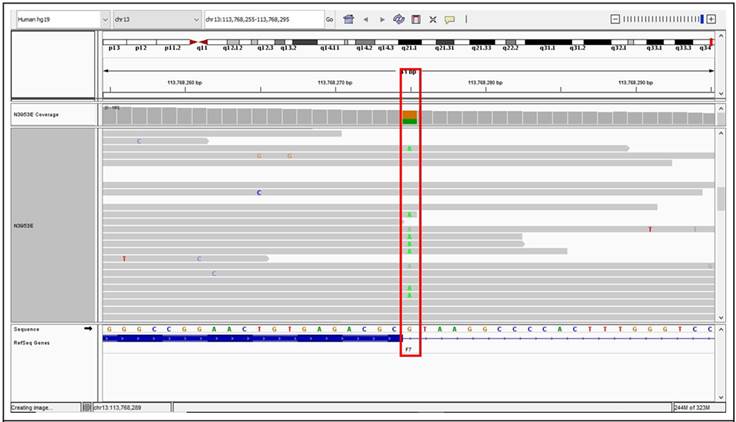
Figura. Imagen en programa IGV en la que se muestra el sitio de splicing entre el exón 5 y el intrón 5 del gen F7. Resaltado en el recuadro rojo, se puede apreciar la posición c.430+1 con una variante en heterocigosis c.430+1G>A
Discusión
La deficiencia congénita de factor VII fue descrita por primera vez en 1951; el factor VII fue secuenciado por completo hasta 1987.8,11,12 La asociación entre las mutaciones en el gen relacionado con el factor VII y las manifestaciones clínicas aún se encuentra en estudio y se cree que existen factores ambientales y hereditarios que modulan la expresividad de la deficiencia y generan discordancia entre el genotipo y el fenotipo de la enfermedad.6,12,13
Los sangrados anormales, en especial en el periodo neonatal, deben obligar al médico estudiado, en que el paciente presenta sangrado umbilical anormal desde el nacimiento que obliga a la madre a consultar en varias ocasiones y, aún más importante, con el antecedente de un hermano que fallece en periodo neonatal por un sangrado cerebral. Para realizar el diagnóstico, se debe efectuar un análisis de rutina (TP, TTP y recuento de plaquetas), seguido de la medición de la actividad coagulante del FVII. Es necesario repetir la prueba del FVII al menos una vez para confirmar la deficiencia; adicionalmente, aunque no es indispensable, resulta útil llevar a cabo estudios de otros factores de coagulación dependientes de vitamina K para descartar deficiencias combinadas, de ser este el caso, el TTP se encontraría prolongado también. 9,12,14 Como se puede ver en el Cuadro, este paciente presentó alteración en el TP aislada con TTP normal y fibrinógeno normal, lo cual establece la sospecha diagnostica de deficiencia congénita de factor VII, confirmado por la cuantificación del mismo factor.
Cuadro Tiempos de coagulación y factor VII del paciente al ingreso y posterior a profilaxis
| Estudios de laboratorio | Al Ingreso | Posterior a dosis de factor VII a las 12 h | En profilaxis con rFVIIa | Referencia |
|---|---|---|---|---|
| TP s | 158.4 | 24% | Menos de 10 | 13.5-16.4 |
| Porcentaje de actividad TP% | 005.0 | 42% | 100 | 70-100 |
| TTP s | 033.1 | 30% | 029 | 31.2-44 |
| Factor VII% | menos de 1 | 14% | 091 | 67-107 |
s=segundos
Este padecimiento presenta manifestaciones clínicas no severas y leves. Los pacientes sintomáticos se pueden dividir en aquellos con tendencia a sangrados de leves a moderados, los cuales usual- mente son mucocutáneos, al igual que ocurre en los trastornos plaquetarios, y aquellos que presentan riesgo de desarrollar hemorragias que potencial- mente arriesguen su vida o sus extremidades10,15. El sangrado del cordón umbilical está asociado a un alto riesgo de desarrollar hemorragias más graves, a nivel de sistema nervioso central y gastrointestinal, estos sangrados usualmente ocurren poco después del nacimiento. El paciente de este caso clínico pre- sentó sangrado de cordón umbilical que efectiva- mente lo predispuso a hemorragias más graves como la del SNC, la cual presentó a los 2 meses de nacido; ambos cuadros clínicos son altamente sugestivos de deficiencia de factor VII. Otros sangrados que pue- den generar sospecha de una deficiencia de FVII incluyen sangrado después de la circuncisión o de la punción de talón durante el periodo neonatal y las extracciones dentales en la infancia.12,15
El objetivo de un diagnóstico temprano y específico de la deficiencia de algún factor de la coagulación es la administración de la terapia especifica. Para el caso en particular de deficiencia congénita de factor VII, existen varias opciones terapéuticas: rFVIIa, factor VII derivado de plasma, PFC y concentrados de complejo de protrombina. El rFVIIa es la terapia de reemplazo más utilizada, su ventaja principal es la eficacia, además de no ser un derivado de plasma humano; sin embargo, el costo por dosis es muy elevado.La indicación más frecuente para la profilaxis a largo plazo son las hemorragias a nivel de sistema nervioso central, seguidas de hemartrosis y sangrado gastrointestinal, esta debe haber iniciado poco después del primer sangrado significativo en las formas severas de deficiencia de FVII, como le ocurrió al paciente que aquí se describe. Los sangrados menos graves no requieren terapia de reemplazo recombinante y suelen resolverse con agentes hemostáticos locales y solo administra terapia de reemplazo en la preparación de eventos quirúrgicos.12 En este caso, el paciente se encuentra recibiendo profilaxis 5 días de la semana con rFVIIa para el manejo de su desorden.
La deficiencia de factor VII es el desorden congénito más común y tiene un amplio rango de manifestaciones clínicas que van desde sangrados leves hasta graves. El diagnóstico se sospecha por la alteración de las pruebas de tiempos de coagulación y se confirma con la medición del factor VII. Se debe sospechar en familias consanguíneas o con historial de sangrados graves. La confirmación molecular e identificación de la mutación de F7 ayuda a establecer un diagnóstico claro y definitivo para el inicio de una terapia temprana y consejo genético.