Enfermedad por Hemoglobina H; primer caso de la variante de hemoglobina H tipo (-α3.7/ --SEA) en Costa Rica.
(Hemoglobin H Disease; First Case of Hemoglobin H Variant Type (-α<3.7/ --SEA) in Costa Rica)
Walter Cartín-Sánchez1*, Kathia Valverde-Muñoz2, Carlos Suárez-Vargas1, Jorge López-Villegas11. Laboratorio de Estudios Especializados e Investigación. 2. Servicio de Hematología. Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”, San José, Costa Rica. ]]>
Abreviaturas:Hb,hemoglobina;HbA:hemoglobinaA;Hb A2,hemoglobina A2; HCM, hemoglobina corpuscular media; Hb F, hemoglobina fetal; Hb H, Hemoglobina H; HPLC, cromatografía líquida de alta presión; Hto, Hematocrito; PCR: reacción en cadena de la polimerasa; VCM: volumen corpuscular medio. Correspondencia: Walter Cartín. Correo electrónico: walter.cartin@gmail.com Resumen: La enfermedad por Hemoglobina H es la forma más común de talasemia intermedia y posee muchas características que requieren cuidadosa consideración en su manejo clínico. En la mayoría de los casos, la enfermedad por Hemoglobina H resulta de un estado doble heterocigoto producido por una deleción tipo α0 que remueve ambos genes de α-globina en uno de los cromosoma 16 y de una deleción tipo α+ en uno de los genes de α-globina en el otro cromosoma 16, resultando en una condición tipo (--/-α). El exceso de cadenas β de globina precipita y forma una hemoglobina anormal característica; la hemoglobina H (Hb H), un tetrámero de β globina (β4). Los pacientes con hemoglobina H que se encuentran en estado compensado pueden tener niveles de hemoglobina entre 9 y 10 g/dL, sin embargo durante las crisis hemolíticas, que se desarrollan durante o después de infecciones agudas con fiebres altas, la hemoglobina puede llegar a disminuir significativamente y los pacientes pueden desarrollar shock y fallo renal.Aún cuando la esplenectomía eleva la hemoglobina significativamente, no se recomienda porque la mayoría de los pacientes tienen un nivel aceptable de hemoglobina mientras se encuentren compensados. Se presenta el primer caso descrito en Costa Rica de enfermedad por hemoglobina H variante del sudeste asiático (-α3.7/ --SEA).1 Descriptores: hemoglobina H, alfa talasemia, anemia hemolítica.
]]>
Abstract: Hemoglobin H (Hb H) disease is the most common form of thalassemia intermedia and has many features that require careful consideration in its management. In the majority of cases, the disease results from double heterozygosity for α0thalassemia due to deletions that remove both linked αglobin genes on one chromosome 16, and deletional α+ from single α-globin gene deletions on the other chromosome 16 resulting in a (--/-α) condition. The excess β globin chain precipitates and forms a characteristic abnormal hemoglobin: hemoglobin H a β globin tetramer (β4). In a steady state, patients with Hb H disease have hemoglobin levels around 9 to 10 g/dL however, during a hemolytic crisis, which frequently occur in or after acute infections causing high fever, the hemoglobin may drop significantly and the patients can develop shock or renal shutdown. Even though splenectomy leads to significant elevation of hemoglobin levels, it is not recommended because the majority of patients do well with said steady-state hemoglobin levels. We present here the first case of hemoglobin H (-α3.7/ --SEA) southeast Asia variant described in Costa Rica.1 Keywords: hemoglobin H, alpha-thalassemia, hemolytic anemia.
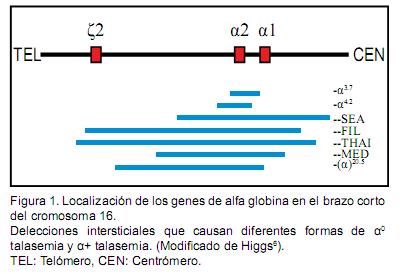
En individuos normales existen un par de genes ligados de α-globina (αα) localizados en cada cromosoma 16 (16p13.3), por lo tanto posee 4 genes de alfa globina (αα/αα) y se designa gen α-2 a la copia más telomérica y gen α-1 al más adyacente al centrómero. Las alfa talasemias son un grupo de anemias hereditarias asociadas a un déficit variable de cadenas de α-globina que refleja el número de genes de α-globina afectados y se divide en dos tipos: delecional y no delecional. El tamaño de la deleción es importante y afecta el fenotipo clínico (Figura 1). Son de los desórdenes genéticos más comunes ya que afectan al 5% de la población mundial; siendo todavía más frecuente en el sudeste asiático (≈40% población).2,3
Se han descrito un poco más de 40 mutaciones delecionales diferentes, de las cuales las más frecuentes son las de deleción simple: la deleción-α,4.2y la deleción –α,3.7 2 De las deleciones complejas, la más frecuente es la --SEA., en la cual hay una deleción de ambos genes de α-globina en un cromosoma (genes en posición cis).2 ]]>
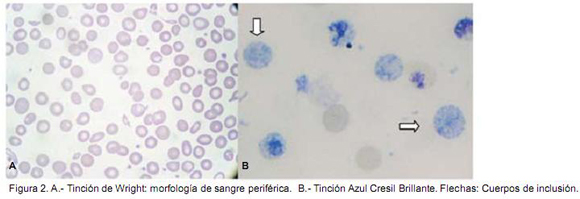
La clínica de las alfa talasemias es muy variable. En un extremo se encuentra la condición completamente asintomática que resulta de la disfunción en uno de los cuatro genes de αglobina (-α/αα) lo que se conoce como rasgo talasémico. En contraste, la deleción de los cuatro genes de α-globina (--/--) resulta en una enfermedad in-uterofatal, conocida como síndrome hydropsfetalispor hemoglobina Bart. Las alfa talasemias se pueden clasificar de acuerdo a la severidad de la pérdida de las cadenas alfa, así cuando hay una pérdida completa de producción de la cadenas de α-globina en un mismo cromosoma se tiene lo que se conoce como α0-talasemia. Por otro lado las α+-talasemias se caracterizan por una reducción en la producción de α-globina, la cual puede ser causada ya sea por mecanismos delecionales o no delecionales de un gen de α-globina. Los dobles heterocigotos para α0-talasemia y α+talasemia (--/-α) solo tienen un gen funcional de alfa globina y presentan por lo tanto un desbalance severo en la síntesis de cadenas de α-globina. El exceso de cadenas de β-globina precipita y forma una hemoglobina anormal característica, la hemoglobina H (Hb H) que es un tetrámero de β-globina (β4). La severidad clínica va a depender de si la condición es secundaria a una mutación de tipo delecional o no.4, 5 Los fenotipos clínicos de la enfermedad por Hb H que ocurren por mecanismos no delecionales son generalmente más severos que los causados por el tipo deleccional, esto se ve en los pacientes con Hb H del tipo delecional, los cuales tienen un desarrollo y crecimiento normal, como es el caso de nuestra paciente. Las pruebas de laboratorio para el diagnóstico de enfermedad por Hb H incluyen: el hemograma, la electroforesis de hemoglobina, HPLC y el análisis del ADN genómico. Hallazgos típicos en el hemograma de las talasemias (incluyendo a las alfa talasemias) son la eritrocitosis con anemia, índices de glóbulos rojos disminuidos (VCM, HCM) y un aumento en el RDW, lo que se refleja en el frotis de sangre periférica con una morfología de glóbulos rojos con marcada hipocromía y anisopoiquilocitosis (Figura 2A). Las inclusiones en los glóbulos rojos por Hemoglobina H se pueden demostrar luego de la incubación de sangre periférica con colorantes supravitales (Figura 2B). El HPLC permite cuantificar los diferentes tipos de hemoglobinas incluyendo la Hb H, la cual se confirma mediante métodos moleculares.
]]>
Caso Clínico
La paciente fue referida al servicio de Hematología del Hospital Nacional de Niños Dr. Carlos Sáenz Herrera, San José Costa Rica a la edad de 4 años, con historia de presentar anemia desde el año de edad y sin respuesta al tratamiento con hierro oral. La niña hasta el momento de la referencia no presentaba ningún síntoma, pero se le documentaba disminución en el nivel de hemoglobina por lo cual seguía un control en la clínica de su localidad. El único antecedente familiar de importancia, con el cuadro presente, es el diagnóstico de drepanocitosis en una prima. El hemograma al momento de la referencia mostró un cómputo de glóbulos rojos de 5.93 millones/μL, una Hb: 9.8 g/dL, VCM: 57 fL, HCM: 16.1 pg, un RDW: 20% y plaquetas y fórmula blanca normales. Se decidió realizar índices de hierro, los cuales se encontraron dentro del rango de referencia (50-120μg/dL) por lo cual se considera la posibilidad de una hemoglobinopatía. Se le realiza electroforesis de Hb que demuestra una HbA2en 1.2% y una Hb Fetal en 1.7% lo que descarta los diagnósticos de beta o delta-beta talasemia. Sin embargo, el paciente mantenía niveles relaltivamente bajos de Hb y hemólisis persistente lo que se traducia clínicamente como ictericia en escleras y elevación de los niveles de bilirrubina indirecta con disminución constante de los niveles de hemoglobina, lo cual motivó a considerar otro tipo de hemoglobinopatía. Se solicitó: -cuerpos de inclusión, el cual resultó positivo; - HPLC (BioRadVariant II), el cual reveló: Hb A: 97%; Hb A2: 1.9% y Hb F: 0.6%. Adicionalmente se observó una pequeña fracción de eluido con un tiempo de 0.5 minutos consistente con Hb H.
El ADN genómico se extrajo de leucocitos de sangre periférica. Se llevaron a cabo estudios utlizando la técnica de PCR con primers diseñados para detectar tanto la deleción simple del gen de α-globina del tipo -α3.7 como la de tipo –α4.2 y la deleción compleja --SEA. Por los resultados obtenidos en esas determinaciones, se concluye que la paciente tiene enfermedad por Hb H con deleción de tres genes de α-globina, dando un genotipo -α3.7/--SEA. El resto del gen de α-globina fue amplificado por PCR, los productos purificados y se realizó la secuencia de nucleótidos. No se encontraron mutaciones del tipo No delecional en la cadena de α-globina.
Discusión Generalmente los pacientes con enfermedad por Hb H tienen valores de hemoglobina entre 8 y 9 g/dL y no necesitan de transfusiones regulares o terapia por quelación de hierro. Se recomienda suplementar la dieta con acido fólico (entre 2 y 5 mg/día), especialmente en pacientes pediátricos que pueden no estar adquiriendo suficientes nutrientes de la dieta diaria debido al aumento de los requerimiento por la eritropoyesis aumentada. ]]>
En las crisis hemolíticas, las cuales son frecuentes durante o después de una infección aguda o por fiebres altas, los valores de hemoglobina pueden disminuir en una noche hasta niveles de 3 g/dL, debido a que los eritrocitos que contienen Hb H precipitada en su interior son destruidos rápidamente. La crisis aguda puede ser tan severa como la que se observa en pacientes con deficiencia de G6PD.2 Los pacientes pueden caer en shock con fallo renal agudo. Esta es probablemente la única complicación mayor en la enfermedad por Hb H que requiere intervención inmediata. Cuando esto sucede se debe administrar rápidamente transfusiones sanguíneas y antibiótico terapia. En el cuadro numero uno se resumen las guías para el manejo de las crisis hemolíticas que se presentan en la enfermedad por Hb H.
Aún cuando la esplenectomía puede elevar significativamente los niveles de hemoglobina en la enfermedad por Hb H, generalmente no se recomienda debido a que la mayoría de los pacientes manejan niveles adecuados de hemoglobina cuando se encuentran compensados. Algunas medidas preventivas tales como vacunación por neumococo y dosis bajas de aspirina profiláctica en los casos con trombocitosis post esplénica (cómputo de plaquetas > 1,000,000/uL) deben de considerarse para evitar las consecuencias de la post esplenectomía tales como las infecciones bacterianas y la trombosis venosa. Los episodios hemolíticos agudos son más comunes en la primera década de vida (debido a infecciones por inmadurez del sistema inmune) y pueden contribuir en la severidad clínica de pacientes con Hb H. Pareciera que la hemólisis aguda disminuye su frecuencia al inicio de la segunda década de vida, y la mayoría de los pacientes no requieren más transfusiones para cuando alcanzan la pubertad3. Se ha sugerido que los episodios de anemia hemolítica están asociados y parcialmente explicados por infecciones recurrentes y elevación de la temperatura. Adicionalmente los episodios de infecciones recurrentes pueden resultar en una hiperfunción del bazo, y esta expansión del bazo debido a las infecciones, puede atrapar más glóbulos rojos y deteriorar aún más la condición anémica. ]]>
Este es el primer caso de enfermedad por Hb H variante del sudeste asiático (-α3.7/--SEA) que se diagnostica en Costa Rica1. Actualmente la paciente se encuentra estable, y se está realizando un estudio familiar para brindar el consejo genético correspondiente a fin de evitar complicaciones tan serias como el síndrome hydropsfetalispor hemoglobina Bart, una condición intrauterina fatal.6
Recibido: 8 de marzo de 2010 Aceptado: 13 de abril de 2010
Referencias
1. Sáenz GF, Rodríguez W. Síndromes talasémicos. Nuevos conceptos y estado actual del conocimiento en Costa Rica. Act Méd Costarric 2006; 48:4. [ Links ] 2. Fuchaeron S, Viprakasit V. Hb H disease: clinical course and disease modifiers. American Society of Hematology. Education Program Book. New Orleans, Luisiana. 2009; 26-34. [ Links ] 3. Chui DH, Waye JS. Hydrops fetalis caused by alpha-thalassemia: an emerging health care problem. Blood 1998; 91:2213-2222. [ Links ] ]]>
4. Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood 2003; 101:791-800. [ Links ]
5. Lau YL, Chan LC, Chan YY. Prevalence and genotypes of alpha- and beta-thalassemia carriers in Hong Kong – implications for population screening. N Engl J Med 1997; 336:1298-1301. [ Links ] 6. Higgs DR. Molecular basis of alpha thalassemia. In: Steinberg MH, Forget BG, Higgs DR, Weatheall DJ. DisordersofHemoglobin. Sec. Ed. CambridgeUniversityPress, 2009: 241-265. [ Links ]