Síndrome de Marfán.
Gerardo Vasquez Torres, Oswaldo Gutiérrez Sotelo
Resumen
Se presenta el caso de una familia con síndrome de Marfan diagnosticada a través de uno de sus miembros quien ingresó a la sala de urgencias por un episodio de insuficiencia cardíaca.
Abstract
We present a case of a family with Marfan´s syndrome first diagnosed through one family member who presented to the emergency room with heart failure.
Introducción
El síndrome de Marfán es un trastorno multisistémico del tejido conectivo de transmisión autosómica dominante. Afecta aproximadamente 1 de cada 5000 individuos, aunque se cree que su prevalencia es mayor. Esta condición no muestra una predilección por una raza o zona geográfica en particular. En alrededor de un 25% de los pacientes no existe historia familiar de esta afección, la cual entonces ocurre por la aparición de mutaciones espontáneas. Esta enfermedad tiene un carácter pleiotrópico con manifestaciones en distintos sistemas, especialmente musculoesquelético, cardiovascular y ocular (1,2).
Se ha determinado que la mutación ocurre en el cromosoma 15q21 el cual codifica a una glicoproteína grande llamada fibrilina-1, el componente principal de las microfibras extracelulares que se encuentran en grandes cantidades en el tejido conectivo (1,2).
]]> El diagnóstico es clínico y depende de la combinación de criterios mayores y menores (tabla 1), aunque puede implicar un reto dada la gran variabilidad fenotípica entre los individuos afectados en una misma familia, la baja especificidad de muchos de los signos clínicos y la existencia de otros desórdenes genéticos microfibrilares similares a este síndrome tales como el síndrome de Ehlers-Danlos, osteogénesis imperfecta, síndrome de Turner o de Noonan (3), ectasia anuloaórtica familiar con disección (7) y el síndrome de aracnodactilia contractural congénita (1).El pronóstico está determinado por la dilatación progresiva de la aorta, la cual potencialmente puede derivar en disección de aorta y muerte en sujetos jóvenes (2,5,7).
Caso clínico
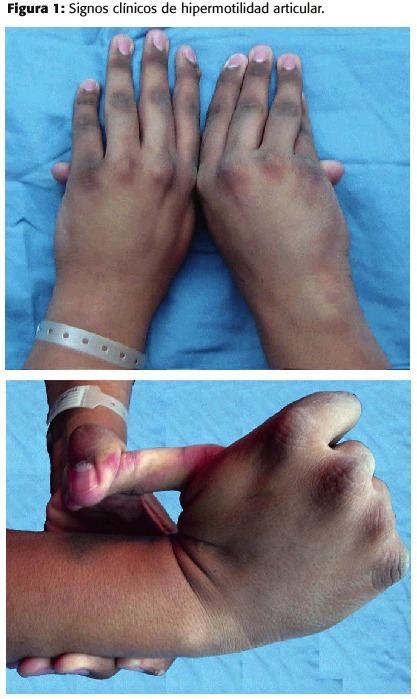
Paciente masculino de 17 años de edad que consulta al servicio de urgencias por presentar en las últimas 3 semanas disnea de moderados esfuerzos que progresó hasta ser de reposo y paroxística nocturna, manejándose inicialmente como insuficiencia cardíaca por miocardiopatía dilatada de causa desconocida. Se le inició terapia de bloqueo neurohormonal con enalapril (inhibidor de la enzima conversora de angiotensina), digoxina, furosemida (diurético de asa), reposo y oxigenoterapia. En término de 72 horas mejoró sustancialmente, desapareciendo el cuadro de disfunción ventricular sistólica.
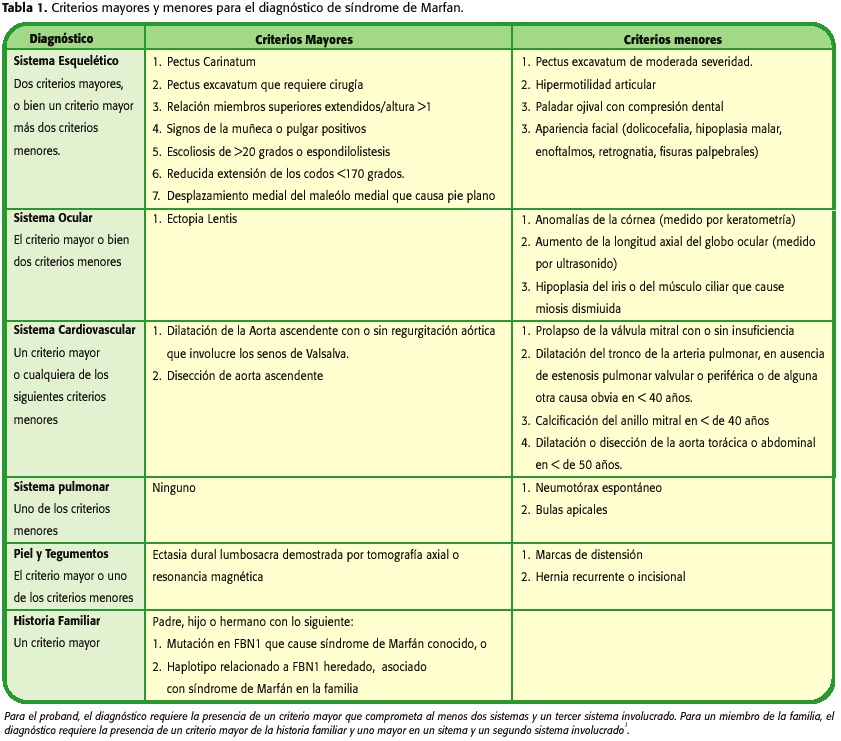
Fue trasladado al servicio de cardiología. El examen físico mostró talla=1.94 m, peso=105 kg, fascies característica del síndrome de Marfán, dolicocefalia e hipoplasia malar; ingurgitación yugular a 45 grados, pulso carotídeo saltón, pulmones con murmullo vesicular disminuido y presencia de crépitos en ambas bases. La semiología cardíaca mostró impulso precordial hiperdinámico y soplo diastólico en el foco aórtico de intensidad III/IV, compatible con insuficiencia aórtica significativa y presencia de tercer ruido. El abdomen presentaba múltiples estrías, era blando, depresible, no doloroso, con hepatomegalia a 3 cm bajo el reborde costal derecho y las extremidades con edema bipodálico moderado. En el sistema musculoesquelético se encontró escoliosis lumbar e hiperelasticidad de articulaciones (figura 1).
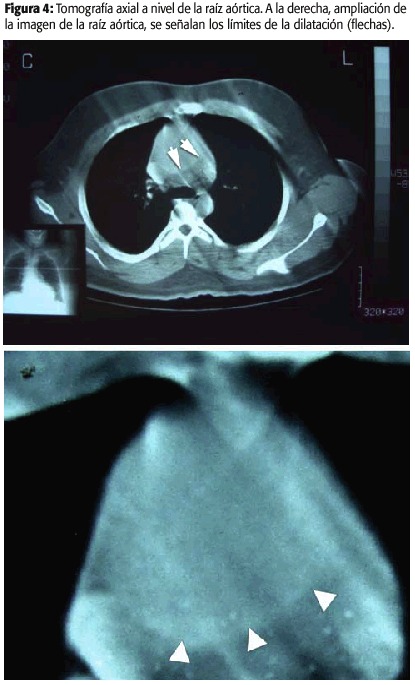
La tomografía axial computarizada fue reportada inicialmente como normal, sin embargo en la figura 4 se nota la presencia de un aneurisma localizado en la aorta ascendente, de dimensiones 7,2 cm por 4,2 cm sin evidencia de disección.
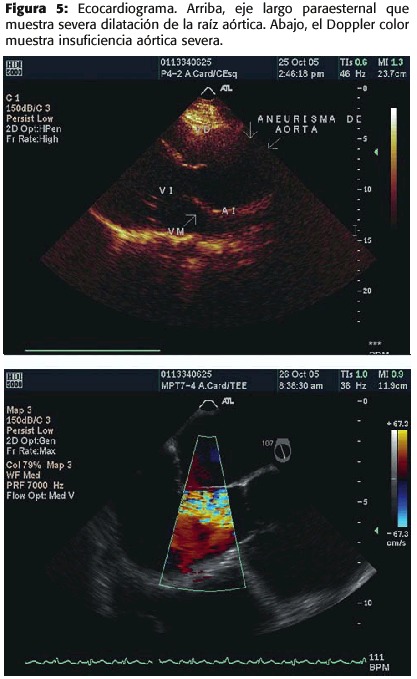
En el ecocardiograma transtorácico, se encontró el ventrículo izquierdo dilatado y con hipertrofia excéntrica, la contractilidad global estaba deprimida en grado moderado a severo con patrón de hipoquinesia difusa y fracción de eyección del 30%. La aorta presentaba importante dilatación de la raíz (figura 5,A). Al interrogatorio Doppler se documentó regurgitación aórtica severa con tiempo de hemipresión de 150 ms (figura 5,B). El ecocardiograma trans-esofágico confirmó la presencia del aneurisma de la raíz de aorta ascendente limitado a esta zona y ausencia de disección.
]]>
El paciente fue referido al servicio de cirugía cardiovascular para la corrección de su aneurisma y colocación de una prótesis valvular aórtica mecánica. Se valoró a los familiares más próximos encontrándose un hermano de 21 años con 1,91 m de estatura también con características fenotípicas del síndrome y la raíz de la aorta en 47 mm, una hermana de 13 años con 1,81 m de estatura, también con similares características y raíz de aorta en 38 mm y una niña de 10 años de 1,65 m de estatura con raíz de aorta de 30 mm.
Discusión
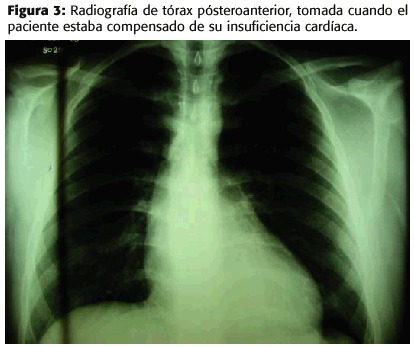
Las características más sobresalientes de este caso son la juventud, el debut con insuficiencia cardíaca y la detección de insuficiencia valvular aórtica en presencia de signos fenotípicos del síndrome de Marfan. Sin embargo, el pedículo vascular normal en la radiografía de tórax y la tomografía axial computarizada del tórax (con limitaciones técnicas) reportada inicialmente como normal podrían haber diferido el diagnóstico más importante: el aneurisma aórtico. Fue con el ecocardiograma que se detectó su presencia, que es el determinante pronóstico en estos pacientes (2).
Este hallazgo es relativamente infrecuente en personas jóvenes. En una serie de 13 pacientes menores de 25 años, los síntomas de presentación, a diferencia de este caso, fueron predominantemente dolor toráccico o datos de isquemia intestinal (3). Se han señalado como factores de riesgo para dilatación aórtica en este grupo etáreo la presencia de condiciones vasculares degenerativas o inflamatorias, cardiopatía congénita, trauma toráccico, uso de cocaína, ejercicio isométrico y gestación (3). En nuestro país se han reportado también casos en la población pediátrica (4).
La mayoría de pacientes en quienes se diagnostica este síndrome tiene ya dilatación de la aorta (60%) (6) y aproximadamente un 10% tiene progresión rápida hacia la dilatación: >1,7 mm/año en mujeres o >1,5 mm en hombres (5). Los factores asociados a progresión rápida son mayor edad, hipertensión arterial, diámetro aórtico inicial, insuficiencia aórtica significativa y patrón de llenado diastólico anormal por Doppler (5).
Es relevante comentar que este fue un diagnóstico tardío, pese a que este paciente presentaba características esqueléticas que obligan a sospechar la enfermedad y valorar a los familiares. Es inusual en nuestras latitudes encontrar personas con estaturas como las encontradas en esta familia, hallazgo que debe ser el indicio para la búsqueda de otros criterios que nos permitan detectar a estos pacientes, que potencialmente están en riesgo de presentar eventos graves como insuficiencia cardíaca, disección aórtica y muerte cardíaca en edades tempranas (2).
Este paciente no presentaba las otras complicaciones cardiovasculares descritas en este síndrome como el prolapso de la válvula mitral, dilatación del tronco de la raíz de la arteria pulmonar (el criterios es >34,8 mm) (2), calcificación del anillo mitral o compromiso de la aorta distal o la abdominal.
En conclusión, este caso demuestra la importancia de la sospecha clínica ante los hallazgos semiológicos descritos, de la detección de familiares afectados y la vigilancia e intervención temprana para la modificación del pronóstico (7).
Referencias
]]>1. Ho NCY, Tran J, Bektas A. Marfans síndrome. Lancet 2005; 3: 1978-81 [ Links ]
2. Nollen GJ, Mulder BJM. What is new in the Marfan syndrome? International Journal of Cardiology 2004. 97: 103-108 [ Links ]
3. Zalzstein E, Hamilton R, Zucker N, Diamant S, Webb G. Aortic dissection in children and young adults: diagnosis, patients at risk, and outcomes. Cardiol Young 2003; 13: 341-344 [ Links ]
4. Mas C, Iturrino R. Protocolo de manejo cardiovascular en el síndrome de Marfán. Revista Costarricense de Cardiología 2004; 6 (3) : 39-42 [ Links ]
5. Meijboom LJ, Timmermans J, Zwinderman AH, Engelfriet PM, Mulder BJM. Aortic Root Growth in Men and Women With the Marfans Syndrome. Am J Cardiol 2005; 96: 1441-4 [ Links ]
6. Lazarevic AM, Nakatani S, Okita Y, Marinkovic J, Takeda Y, Hirooka K, Matsuo H, Kitamura s, Yamagishi m, Miyatake k. Determinants of rapid progression of aortic root dilatation and complications in Marfan syndrome. International Journal of Cardiology 2006; 106: 177-82 [ Links ]
7. Nienaber C, Eagle K. Aortic Dissection: New Frontiers in Diagnosis and Management. Part I. From Etiology to diagnostic Strategies. Circulation 2003, 108: 628-635. [ Links ]
]]>
Servicio de cardiología, Hospital México, C.C.S.S., San José, Costa Rica.
Servicio de cardiología, sección 4-B, Hospital México, San José, Costa Rica. Teléfono (506) 242-6646; Fax (506) 231-3856 e-mail oswcr@hotmail.com
]]>