Sitio de realización. Instituto de Genética Humana, Universidad Friedrich Alexander Erlangen-Nuremberg e INISA, Universidad de Costa Rica.
Materiales y métodos. Se obtuvo ADN de 19 personas emparentadas por vía materna con un paciente afectado con DMD; para cada uno se analizaron, mediante PCR, 5 marcadores microsatelíticos ubicados en la región del gen de la distrofina. Se marcó un imprimador de cada par con un fluorocromo, se determinó el tamaño de los productos de amplificación por electroforesis capilar fluorescente y se construyeron haplotipos para la región de interés.
Resultados. Se determinó el haplotipo de riesgo en el paciente, el que también se encontró en dos primos suyos, adultos sanos y en tres de sus tías y tres primas. Los hermanos del paciente heredaron de su madre el cromosoma X portador del haplotipo normal. ]]>
Conclusiones. Los resultados indican que el paciente heredó una mutación de novo, originada en la línea germinal ya sea de la madre o de la abuela materna y que ninguna de las otras mujeres de la familia está en riesgo incrementado de ser portadoras. No es posible determinar si la madre del niño es portadora de la mutación.Palabras clave: distrofinopatía, herencia ligada al X, PCR, electroforesis capilar fluorescente, haplotipos, microsatélites
La distrofia muscular de Duchenne (DMD) (1,2) y la distrofia muscular de Becker-Kiener (BMD) (3) representan en conjunto más del 40% de las distrofias musculares (4). Ambas enfermedades son alélicas, es decir causadas por mutaciones en el mismo gen, el de la distrofina, y se conocen como distrofinopatías (5). El gen de la distrofina es el más grande que se conoce hasta el momento; tiene al menos 79 exones y abarca 2.4 megabases (6) en el brazo corto del cromosoma X (7). La herencia de estas enfermedades es recesiva ligada al cromosoma X (8,9). Esto quiere decir que en la gran mayoría de los casos los afectados son hombres ya que poseen una sola copia del cromosoma X, mientras que las mujeres con mutaciones son por lo general portadoras. Aunque ambas enfermedades se caracterizan por un debilitamiento progresivo de los músculos para el cual no hay tratamiento efectivo, la progresión de la DMD es mucho más rápida y su desenlace es fatal, falleciendo los pacientes alrededor del inicio de la segunda década de vida, mientras que los pacientes con BMD alcanza la edad adulta y se conocen casos en que llegan a reproducirse; además la DMD tiene una mayor incidencia (1:3500 niños varones nacidos vivos) que la BMD (1:18500 niños varones nacidos vivos) (4).
Cerca de dos tercios de las mutaciones causantes de las distrofinopatías son deleciones de uno o más exones (10). Del 5 al 10% de los casos son causados por duplicaciones (11). El resto se debe a mutaciones de punto (12,13) como son sustituciones de bases y pequeñas inserciones o deleciones, así como a inversiones (14). Se ha observado que el fenotipo no está correlacionado con el tamaño de las mutaciones sino con el efecto que estas tengan sobre el marco de lectura del gen (15). Las mutaciones que conservan el marco de lectura, esto es, que aunque se pierda parte del gen el resto mantiene la secuencia original, causan por lo general BMD. En muchos casos de BMD las mutaciones originan la pérdida o cambio de algunos aminoácidos, por lo que la distrofina es parcialmente funcional. Las mutaciones que alteran el marco de lectura resultan en ausencia total de proteína, en una proteína severamente truncada o muy diferente de la distrofina, que no es funcional y por lo tanto se asocian con el fenotipo severo de la DMD, aunque existen excepciones (16). En la actualidad es posible detectar la mayoría de las deleciones y duplicaciones grandes por medio de la reacción en cadena de la polimerasa (PCR)-múltiplex (17).
Debido a que la DMD es una enfermedad letal ligada al cromosoma X y a que la tasa de mutación del gen de la distrofina es alta (18,19), se estima que una tercera parte de los casos de DMD se debe a mutaciones de nava, es decir, la mutación se origina en la línea germinal de la madre, pero la madre no es una portadora de la enfermedad. Para efectos de consejo genético es de gran importancia poder distinguir si las mujeres emparentadas en primer grado con pacientes con DMD son o no portadoras de la mutación.
Cuando un paciente tiene una deleción o duplicación grande es relativamente sencillo determinar si las mujeres de la familia emparentadas en primer grado con él son o no portadoras de la mutación. Para esto se puede utilizar PCR-múltiplex cuantitativo (20,21,22) en el que se comparan dosis génicas entre mujeres control y posibles portadoras. También es posible la detección de portadoras con técnicas citogenéticas como FISH (hibridación in situ fluorescente) (23).
Para el 35% de los pacientes con DMD que tienen mutaciones de punto es mucho más difícil determinar cual es la mutación que poseen. Es necesario recordar que en estos casos se está buscando lo que posiblemente sea el cambio de una base por otra en 79 exones que abarcan 2.4 megabases. Además, no existen regiones dentro del gen en las cuales la incidencia de este tipo de mutaciones sea más alta, ni tampoco existe una o varias mutaciones que tengan una más alta frecuencia (13). Es decir, cuando se quiere localizar una mutación de punto se debe analizar todo el gen, por lo que dado su enorme tamaño la secuenciación no es una opción viable. Otras técnicas como polimorfismos de conformación en simple banda (SSCP) múltiplex (24), cromatografía líquida desnaturalizante de alto desempeño (DHPLC) (25), electroforesis desnaturalizante en gel con gradiente de concentración (DGGE) (26), prueba del truncamiento de proteínas (PTT) (27), entre otras, se han utilizado con ese propósito. Todas ellas consumen mucho tiempo y dinero, además de que no ofrecen absoluta certeza de que detecten la mutación, por lo que los laboratorios no ofrecen diagnóstico directo a pacientes con mutaciones de punto.
El diagnóstico en estos casos se puede hacer en forma indirecta tratando de identificar al cromosoma portador de la mutación. Para ello se caracteriza la variación en distintas regiones, llamadas marcadores genéticos, alrededor y dentro del gen, esto es, construyendo haplotipos de la región (28). Si el marcador y el gen están suficientemente cerca el uno del otro se dice que están ligados; es decir, la mayoría de las veces se transmitirán juntos. Según esto, los dos cromosomas X maternos, el portador de la mutación y el portador del alelo normal, deben tener haplotipos diferentes si los marcadores muestran mucha diversidad en la población, por lo que en la actualidad, los marcadores más informativos (porque tienen usualmente varios alelos, cada uno de ellos en frecuencias considerables en la población) son un tipo de marcadores de ADN llamados marcadores microsatelíticos, los cuales están compuestos por secuencias cortas, generalmente de 1 a 4 nucleótidos de longitud que se repiten tras de sí varias veces, en tándem, y cuya posición en el cromosoma, y por lo tanto, con respecto al gen de interés, se conoce. El alelo que presente el individuo afectado (en este caso es solo uno por marcador porque se estudia el cromosoma X) en cada uno de los marcadores empleados sirve para determinar la combinación de alelos en la región de interés, o sea, para establecer el haplotipo. Por lo tanto, se considera a la combinación de alelos en cada uno de los de marcadores que presenta el individuo afectado como el haplotipo de riesgo de tener la mutación. Este tipo de diagnóstico indirecto se puede utilizar tanto para posibles afectados, en diagnóstico prenatal por ejemplo, como para posibles portadoras en familias donde existe al menos un niño con la enfermedad. ]]>
La utilización de estos marcadores se ha facilitado enormemente gracias a la PCR. A partir de las secuencias que flanquean la región donde se encuentra el microsatélite se diseñan imprimadores que amplificarán las repeticiones. Como resultado se observan segmentos de ADN, alelos, de diferente tamaño. Por ejemplo para un marcador tetranucleotídico donde la secuencia que se repite es CAGT una persona podría tener un alelo con 50 repeticiones y en el cromosoma homólogo uno con 55. En este caso al analizar los productos de PCR en un gel se observarán dos bandas que difieren en 20 pares de bases. En la variante que se utilizó en este trabajo, los imprimadores están marcados por moléculas fluorescentes lo que permite mediante electroforesis capilar, determinar el tamaño de los alelos sin necesidad de un gel.En Costa Rica no hay datos sobre la epidemiología de las distrofinopatías. En dos estudios genéticos previos se observó un aparente exceso, según lo esperado, de pacientes cuyos padecimientos no se deben a deleciones (29) así como una elevada proporción de casos debidos a mutaciones de novo (22). En este trabajo se utilizaron 5 marcadores microsatelíticos del cromosoma X para establecer el haplotipo de riesgo en la familia de un paciente con DMD al que no se le detectaron deleciones grandes con el objetivo de determinar el riesgo de ser portadoras que corren mujeres emparentadas en primer grado por línea materna con él.
Materiales y métodos
La familia: El paciente índice presentó los primeros síntomas de distrofinopatía a los 2 años; a los 6 años se le diagnosticó como afectado por DMD por presentar la maniobra de Gowers, seudohipertrofia leve de las pantorrillas y niveles elevados de creatina quinasa sérica (3000-12000 U/I). El paciente tiene en la actualidad 18 años y está en silla de ruedas desde los 11 años. Mediante entrevista con la madre del paciente índice (quien tiene niveles normales de creatina quinasa sérica) se elaboró la genealogía de la familia y se determinó quiénes estarían a riesgo de ser portadoras así como qué otros miembros serían informativos para el análisis genético Se extrajeron muestras de ADN, usando el método de sal (30), de los miembros de la familia a partir de sangre total (Vacutainers con EDTA). Las muestras de todas las personas se obtuvieron con su consentimiento informado o el de sus representantes legales.
Tamizaje de deleciones por PCR-múltiplex:
Se hizo para 28 exones y 2 promotores en 4 reacciones de amplificación independientes según se describió en Sancho-Fernández et al. (29)
Reacciones de amplificación para marcadores microsatelíticos:
Para cada miembro de la familia se realizaron amplificaciones con 5 pares de imprimadores para 5 marcadores dinucleotídicos (repeticiones de 2 pares de bases): DXS1068, DXS 1242, DXS1238, DXS1214 y DXS1202 (31). Se utilizó uno de los imprimadores, (el F) del par correspondiente a cada marcador, marcado con el fluorocromo 6carboxifluoresceína (6-FAM) para los marcadores DXS 1214 y DXS 1242, el fluorocromo tetraclorofluoresceína (TET) para los marcadores DXS 1068 Y DXS 1202, y el fluorocromo hexaclorofluoresceína (HEX) para el marcador DXS 1238.
Para las reacciones de PCR se utilizaron 20 ng de ADN genómico, 2.5 pmol de cada imprimador, Tris-HCI (pH 8.4) 20mM, KCI 50mM, MgCl2 0.75 mM, cada desoxiribonucleótido trifosfato 0.2 mM y 0.35 unidades Taq ADN polimerasa (GIBCO), en un volumen total de 10 µI. El perfil de PCR fue 94°C por 3 min, seguido por dos ciclos de desnaturalización (94°C por 30 seg), hibridación de los imprimadores (61°C por 45 seg), y extensión (68°C por 45 seg), dos ciclos con temperaturas de 94°C-59°C-68°C (para desnaturalización, hibridación, extensión, los tiempos son los mismos), dos ciclos con temperaturas 94°C-57°C-68°C, 31 ciclos con temperaturas 94°C-55°C-68°C y una extensión final a 68°C por 20 min.
Electroforesis capilar: El análisis mediante electroforesis capilar se hizo con un Analizador Genético ABI 310 (Applied Byosistems) utilízando el programa ABI PRISM 310 Genetic Analizer Data Collection versión 1.0.4 de la Corporación Perkin Elmer (1997) para la recolección de datos. ]]>
Para los productos de amplificación de cada marcador se hizo una dilución 1: 10 v/v en agua destilada. Para las electroforesis capilares se tomaron 4 µI de cada dilución de los productos amplificados, 1 µI de marcador de tamaño molecular (TAMRA 500) y 12 µI de formamida desionizada.Resultados
PCR múltiplex: El análisis del ADN del paciente con PCR múltiplex no mostró ninguna deleción, por lo que la determinación del estado de portadoras de mutaciones en el gen de la distrofina en las mujeres emparentadas con él en primer grado por línea materna no se pudo realizar mediante determinación de dosis génica. Esto lleva a que la evaluación se deba realizar de forma indirecta determinando los haplotipos en los cromosomas X que están segregando en esta familia.
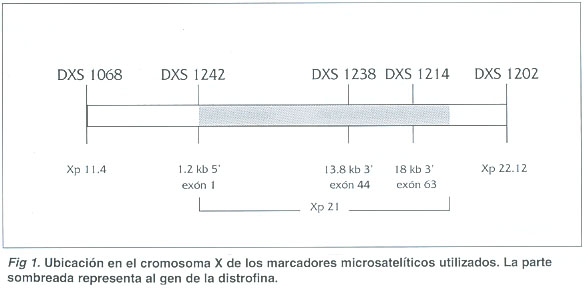
Haplotipos: En la figura 1 se observa la posición de los cinco marcadores utilizados respecto al gen de la distrofina. Dos de los marcadores (DXS1068 y DXS1242) flanquean al gen en el extremo 5', dos marcadores son intragénicos (DXS1238 y DXS1214) y el último (DXS1202) flanquea al gen en el extremo 3'.
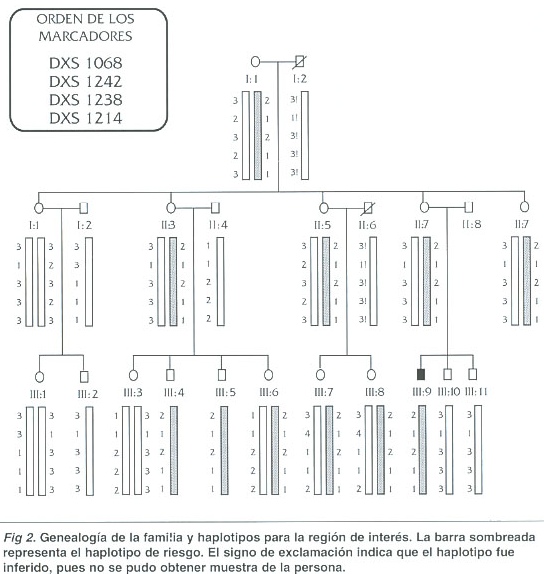
Sin embargo, el análisis del resto de la familia proporciona suficiente información para llegar a conclusiones sobre el posible origen de la mutación y su riesgo de transmisión a otros miembros de la familia. Tres de las tías del niño presentan el haplotipo de riesgo (II:3, II:5, II:9) y una de ellas, la II:3, tiene dos hijos varones con este haplotipo. Estos individuos son adultos sanos, sin ningún síntoma de DMD. Esto indica que en el resto de la familia el haplotipo de riesgo no se asocia con la mutación y sugiere que el paciente sufre de la enfermedad como resultado de una mutación ocurrida en la línea germinal de su madre o de su abuela materna, una mutación de novo. Este resultado sugiere que las mujeres portadoras del haplotipo de riesgo ( I:1, II:3, II:5, II:9, III:6, III:7 y III:8) no son heterocigotas para la mutación en el gen de la distrofina que afecta al paciente.
Los resultados no permiten distinguir, sin embargo, en cuál de las dos posibles mujeres, la madre o la abuela materna, ocurrió la mutación. Si esta ocurrió en la madre, el paciente es el único miembro de la familia con la mutación. Si la mutación ocurrió en la línea germinal de la abuela materna del niño, la madre sería heterocigota. La segregación de los cromosomas X maternos en el núcleo familiar del paciente no permite determinar si esta es la situación, pues los dos hermanos menores del niño afectado recibieron de su madre, por azar, el haplotipo normal y por lo tanto están sanos. En cualquier caso, el destino de la mutación parece haber terminado aquí.
Discusión
Las distrofinopatías son enfermedades particularmente difíciles para el diagnóstico indirecto con cualquier tipo de marcador, incluidos los marcadores microsatelíticos, del estado de portadoras de mutaciones debido al alto porcentaje de recombinación a lo largo del gen, que se estima entre un 10 y un 12% (32). El fenómeno de recombinación, que es un intercambio de segmentos entre parejas de cromosomas homólogos, ocurre durante la meiosis, cuando se están formando los óvulos y los espermatozoides. En el diagnóstico, una recombinación entre el marcador utilizado y el gen de interés puede causar errores, ya que una persona con el alelo de riesgo podría no tener la mutación en el gen de interés. Por lo tanto, se recomienda el uso de marcadores que estén tanto en los flancos del gen como dentro de él. En el presente trabajo se utilizaron tres marcadores que flanquean al gen y dos marcadores intragénicos. En los haplotipos de la familia no se observa ninguna recombinación. Existe la posibilidad de que se haya dado una doble recombinación (dos eventos de recombinación en la región entre dos marcadores) que no se detecta en los haplotipos. Sin embargo, dadas las distancias que separan a los 5 marcadores empleados, la probabilidad de detección de una recombinación es muy alta mientras que la ocurrencia de una doble recombinación es muy baja (33).
Se ha propuesto que para la DMD la mayoría de las deleciones surgen durante la oogénesis y la mayoría de las mutaciones de punto durante la espermatogénesis (34, 35). Sin embargo en este caso la mutación de punto parece haberse originado ya sea en la línea germinal de la abuela o de la madre, por lo tanto durante la oogénesis.
El diagnóstico de portadoras para enfermedades tan severas como la DMD es sumamente importante. Para una mujer heterocigota para una mutación en el gen de la distrofina la probabilidad indica que la mitad de sus hijos varones presentarán la enfermedad y la mitad de sus hijas serán portadoras. Para una mujer no portadora el riesgo de un hijo afectado es igual a la incidencia de la enfermedad en la población.
En este trabajo no es posible determinar si la madre del niño afectado con DMD es o no portadora, pero los datos sí sugieren que el resto de las mujeres de la familia no lo son. Esto es información importante ya que varias de las tías y primas del niño afectado están en edad reproductiva.
En casos donde la DMD es causada por mutaciones de punto, solamente se tiene completa seguridad en el diagnóstico de portadoras cuando se logra identificar la mutación en el niño afectado y se busca en el resto de la familia. Por el momento esta opción no es viable por razones técnicas y económicas. El diagnóstico utilizando marcadores microsatelíticos cuidadosamente escogidos en la región del gen es por ahora, en estos casos, la mejor opción.
Conclusiones ]]>
Los resultados indican que el paciente índice presenta una mutación que ocurrió de novo en la línea germinal ya sea de su madre o de su abuela materna. El resto de las mujeres de la familia no son portadoras, lo cual es información importante para las que están en edad reproductiva, ya que su probabilidad de tener un hijo afectado es muy baja, igual a la incidencia de la enfermedad en la población.Agradecimientos
La estadía de G.C. en Alemania fue posible gracias a una beca del Servicio Alemán de Intercambio Académico (DAAD) y al Instituto de Genética Humana de la Universidad de Erlangen-Nuremberg. El trabajo se financió en parte por la Vicerrectoría de Investigación de la Universidad de Costa Rica, Proyecto 742-97253.
Referencias
1. Duchenne GBA. De I'electrisation localisée et son Application á la Pathologie et á la Thérapeutique. Paris: Bailliére et Fils, 1861. [ Links ]
2. Duchenne GBA. Recherches sur la paralyse musculaire pseudohypertrophique ou paralyse mio-sclerosique. Arch Gén Med 1868; 11 :5-25, 179-209, 305-21, 421-43, 552-88. [ Links ]
3. Becker PE, Kiener F. Eine neue Xchomosomale Muskeldystrophie. Arch Psychiatr Nervenkrankheiten 1955; 193:427-48 [ Links ]
4. Emery AEH. Population frequencies of inherited neuromuscular diseases. A world survey. Neuromusc Disord 1991; 1:19-29. [ Links ]
5. OMIM (TM). Online Mendelian Inheritance in Man. Johns Hopkins University, Baltimore. http://www.ncbi.nlm.nih.gov/omim/ , 2001 [ Links ]
6. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987; 50: 509-17. ]]>
7. Murray JM, Davies KE, Harper PS, Meredith L, Mueller CR, Williamson R. Linkage relationship of a cloned DNA sequence on the short arm of the X chromosome to Duchenne muscular dystrophy. Nature 1982; 300:69-71. [ Links ]
8. Becker PE. Dystrophia musculorum progressiva. Eine genetische und klinische Untersuchung der Muskeldystrophien. Stuttgart: Georg Thieme, 1953 [ Links ]
9. Emery AEH. Duchenne Muscular Dystrophy. Oxford University Press, Great Britain. 1993; 391 p. [ Links ]
10. Gillard EF, Chamberlain JS, Murphy EG, et al. Molecular and phenotypic analysis of patients with deletions within the deletionrich region of the Duchenne muscular dystrophy (DMD) gene. Am J Hum Genet 1989; 45: 507-20. [ Links ]
11. Hu X, Ray P, Murphy G, Thompson M, Worton R. Duplicational mutation at the Duchenne muscular dystrophy locus: its frequency, distribution, origin, and phenotype-genotype correlation. Am J Hum Genet 1990; 46: 682-95. [ Links ]
12. Roberts RG, Bobrow M, Bentley DR. Point mutations in the dystrophin gene. Proc Natl Acad Sci 1992; 89:2331-2335. [ Links ]
13. Prior TW, Bartolo C, Pearl DK, Papp AC, Snyder PJ, Sedra MS, Burgh AH, Mendell JR. Spectrum of small mutations in the dystrophin coding region. Am J Hum Genet 1995; 57:22-33.
14. Baxter PS, Maltby EL, Quarrell O. Xp21 Muscular dystrophy due to X chromosome inversion. Neurology 1997; 49: 260. [ Links ]
15. Koenig M, Beggs AH, Moyer M, et al. The Molecular Basis for Duchenne versus Becker Muscular Dystrophy: Correlation of Severity with Type of Deletion. Am J Hum Genet 1989; 45:498-506. [ Links ]
16. Covone AE, Lerone M, Romeo G. Genotype-phenotype correlation and germline mosaicism in DMD/BMD patients with deletions of the dystrophin gene. Hum Genet 1991; 87:353-360. [ Links ]
17. Chamberlain JS, Gibbs RA, Ranier JE. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucl Acids Res 1988; 16: 11141. [ Links ]
18. Haldane JBS. The rate of spontaneous mutation of a human gene. J Genet 1935; 31:317-326. [ Links ]
19. Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet 1984; 66:17-40. [ Links ]
20. Yau SC, Bobrow M, Mathew CG, Abss SJ. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet 1996; 33: 550-9. [ Links ]
21. loannou P, Chistopoulos G., Panayides K, Kleanthous M, Middleton L. Detection of Duchenne and Becker muscular dystrophy carriers by quantitative multiplex polymerase chain reaction analysis. Neurology 1992; 42:1783-1790. [ Links ]
22. Azofeifa J, Sancho-Fernández VM. Estimación de dosis génica mediante PCR múltiplex y electroforesis capilar fluorescente en posibles portadoras de deleciones en el gen de la distrofina, Costa Rica 1998-2000. Acta Pediátrica Costarricense 2001; 15:64-77. [ Links ]
23. Tocharoentanaphol Ch, Cremer M, Schrock E, et al. Multicolor fluorescence in situ hybridization on metaphase chromosomes and interphase Halo-preparations using cosmid and YAC clones for the simultaneous high resolution mapping of deletions in the dystrophin gene. Hum Genet 1994; 93: 229-35. [ Links ]
24. Kneppers ALJ, Deutz-Terlouw PP, den Dunnen JT, van Ommen GJB, Bakker E. Point Mutation Screening for 16 Exons of the Dystrophin Gene by Multiplex Single-. Strand Conformation Polymorphism Analysis. Human Mutation 1995; 5:232-242. [ Links ]
25. Bennett R, den Dunnen J, O'Brien KF, Darras BT, Kunkel LM. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genetics 2001; 2:17-28. [ Links ]
26. den Dunnen JT, Mulder IM, Koning Gans PAM, Villerius M, van Essen T, van Ommen GB, Buys CHMC, Hofstra R. A whole gene, DGGE-based mutation scan of the dystrophin gene in DMD/BMD patients. American Society of Human Genetics, 1998 meeting. [ Links ]
27. Gardner RJ, Bobrow M, Roberts RG. The identification of point mutations in Duchenne muscular dystrophy patients by using reverse-transcription PCR and the protein truncation test. Am J Hum Genet 1995; 57:311-320. [ Links ]
28. Clemens PR, Fenwick RG, Chamberlain JS, Gibbs RA, de Andrade M, Chakraborty R, Caskey CT. Carrier Detection and Prenatal Diagnosis in Duchenne and Becker Muscular Dystrophy Families, Using Dinucleotide Repeat Polymorphisms. Am J Hum Genet 1991; 49:951-960. [ Links ]
29. Sancho-Fernández VM, Saborío M, de Céspedes C, Azofeifa J. Tamizaje de deleciones en pacientes con distrofina muscular de Duchenne (DMD) o de BeckerKiener (BMD) mediante PCR múltiplex en Costa Rica, 1998-2000. Acta Ped Cost. 2001; 15:78-85. [ Links ]
30. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 1988; 16:1215. [ Links ]
31. Genome Database. Canadá. http://gdb.org , 2001. [ Links ]
32. Abbs S, Roberts RG, Mathew CG, Bentley DR, Bobrow M. Accurate assessment of intragenic recombination frequency within the Duchenne muscular dystrophy gene. Genomics 1990; 7:602-606. [ Links ]
33. The European Molecular Genetics Quality Network. Best practice guidelines Duchenne and Becker Muscular Dystrophy. Inglaterra. http://www.emqn.org . 2001. [ Links ]
34. Grimm T, Meng G, Liechti-Gallati S, Bettecken T, Müller CR, Müller B. On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J Med Genet 1994; 31:183-186. [ Links ]
35. Tuffey S, Chambert S, Bareil C, Sarda P, Coubes C, Echenne B, Demaille J, Claustres M. Mutation analysis of the dystrophin gene in Southern French DMD of BMD families: from Southern blot to protein truncation test. Hum Genet 1998; 102:334-342. [ Links ]
]]>
(*) Biólogos, genetistas, Instituto de Investigaciones en Salud (INISA) y Escuela de Biología, Universidad de Costa Rica(**)Médico, genetista, Instituto de Genética Humana, Universidad Friedrich-Alexander, Erlangen-Nuremberg, República Federal de Alemania
Dirección: Dr. Jorge Azofeifa, Sección Genética Humana, Instituto de Investigaciones en Salud (INISA), Universidad de Costa Rica, San José, Costa Rica. Fax (00506) 207 5130, correo electrónico: azofeifa@biologia.ucr.ac.cr ]]>