Objetivo
Iniciar los estudios genético moleculares sobre las distrofinopatías en Costa Rica. ]]>
Materiales y MétodosTreinta y un pacientes varones, diagnosticados con distrofia muscular, que podrían ser distrofinopáticos fueron reevaluados clínicamente. Veintitrés mostraron un fenotipo de DMD y dos de BMD. Seis no mostraron síntomas definitivos de distrofinopatías. ADN de los pacientes fue analizado mediante PCR-múltiplex en búsqueda de deleciones en el gen de la distrofina. También se realizó un diagnóstico prenatal a una portadora obligada.
Resultados
Diez pacientes presentaron deleciones: en uno abarca 23 exones (del 23 al 25), en dos ocho exones (del 45 al 52 y del 12 al 19), en otro siete exones (del 60 al 66), en dos seis exones (del 45 al 50); en cuatro pacientes la deleción comprende un único exón: el 52 en dos de ellos, el 19 y el 44. Los seis pacientes con diagnóstico dudoso no mostraron deleciones. Un hijo de la portadora obligada, afectado con DMD tiene una deleción de 37 exones (del 6 al 42), mientras que el feto no la tiene.
Conclusión
El traslape de síntomas entre las distintas distrofias musculares, la falta de pruebas diagnósticos de laboratorio y de especialistas en el diagnóstico diferencial evidencian la necesidad de implementar métodos diagnósticos más discriminantes en Costa Rica. La identificación de pacientes con deleciones permite buscar estas mutaciones en las mujeres con posible riesgo de ser portadoras y que así reciban mejor consejo genético.
Palabras clave
distrofia muscular; Duchenne, Becker-Kiener, herencia ligada al cromosoma X, diagnóstico molecular, distrofina, distrofinopatía, deleción, PCR-múltiplex.
]]>
La forma más severa y frecuente es la DMD, padecimiento caracterizado en 1855 por el neurólogo francés Duchenne de Boulogne (1). Su incidencia se estima en 1:3500 recién nacidos varones (3). En un tercio de los casos no hay antecedentes familiares, lo que sugiere que ellos podrían deberse a mutaciones "de novo" (5, 6) reflejándose así una alta inestabilidad del gen. Aunque hay evidencia de que la miopatía ya está presente en el feto, los síntomas se manifiestan entre los 3 y los 5 años como retardo en el desarrollo motor, un caminar de puntillas, seudohipertrofia de los gemelos y maniobra de Gowers (1). La capacidad ambulatorio se pierde alrededor de los 12 años y la muerte sobreviene hacia el final de la segunda década de vida (1).
En 1955 Becker y Kiener (7) describieron, en miembros de la propia familia de Kiener, la forma más benigna y de progresión
más lenta, que hoy lleva su nombre. La incidencia de la BMD se ha estimado en 1: 18 500 recién nacidos varones (3).
En 1982 se demostró que el locus se ubica en el brazo corto del cromosoma X ( Xp21) (8) y en 1985 se identificó el gen (9), lo que permitió comprobar que la DMD y la BMD son desórdenes alélicos. El gen abarca unas 2.4 megabases (2.4 millones de pares de bases) y consta de al menos 79 exones o regiones codificantes (10, 11). Su producto es la distrofina, una proteína de 427 kDa, localizada en la cara interna del sarcolema (12, 13). El gen expresa isoformas particulares en músculo esquelético, liso, cardíaco y en tejido nervioso (12, 13, 14), lo cual indica que puede cumplir varias funciones (15, 16). Estas isoformas se producen por la expresión controlada diferencialmente de al menos 7 promotores específicos de cada tejido y por eventos de edición postranscripcional del ARN mensajero (2, 12,13). Asociado a esto, se ha contemplado recientemente la posibilidad de que una mutación en el promotor de la isoforma cadíaca sea responsable de una cardiomiopatía aislada (17)
Las pruebas diagnosticas muestran biopsias de músculo anormales tanto en la DMD como en la BMD, así como electromiografías alteradas y valores elevados, hasta cien veces sobre lo normal, de creatina quinasa sérica (SCK) (1, 2). Los pacientes con DMD tienen niveles virtualmente indetectables de distrofina, mientras que en aquellos con BMD la proteína se encuentra alterada en tamaño y cantidad, aunque esto no es discriminatorio entre DMD y BMD, ya que se han observado pacientes con niveles sumamente reducidos de distrofina que tienen un fenotipo de BMD (2).
Estudios en varios países han mostrado que entre el 60 y el 65% de los casos de DMD y BMD se deben a deleciones (18, 19, 20). Aunque se han descrito diferentes proporciones en algunos países (21, 22), no hay estudios concluyentes sobre si el patrón de deleciones a lo largo del gen varía con la etnia o la nacionalidad (23). Del 5 al 10% de los casos, con un sobresaliente 14% en Japón (24), presenta duplicaciones (25). El resto se debe a mutaciones de punto (26), y a inserciones e inversiones (27). La mayoría de las deleciones se agrupan alrededor de los exones 3-19 (20%) y 44-52 (80%) (2,1 g), regiones que podrían estar relacionadas con una alta frecuencia de recombinación intragénica ilegítima (28, 29), aunque varios elementos de inserción podrían contribuir a la inestabilidad del gen (30, 31).
Pruebas a nivel de ADN permiten complementar el diagnóstico clínico. Uno de los métodos, la reacción en cadena de la polimerasa (PCR) (32) se implementó para la DMD y la BMD en 1988 por Chamberlain y colaboradores (33) como PCR múltiplex, una variante de la técnica que permite amplificar simultáneamente, empleando una combinación de 18 imprimadores (primers), segmentos de 9 exones del gen de la distrofina en una única reacción (33, 34). Si alguna de las secuencias codificantes está deletada en el gen del paciente, no se obtiene producto de amplificación. Beggs et al. (35) complementaron el protocolo anterior con una combinación de 9 exones adicionales; de esta manera con solo dos reacciones se pueden detectar al menos el 98% de las deleciones responsables de estas enfermedades.
En Costa Rica el diagnóstico clínico de las distrofias musculares rara vez sugiere el tipo diagnosticado, y sólo en una minoría de los casos se cuenta con datos sobre niveles de SCK y electromiografía. Sumado a esto, la falta de documentación sobre la historia familiar, o si esta es negativa, dificulta aún más el diagnóstico correcto. Por lo tanto, las condiciones no han sido propicias para realizar estudios sobre las distrofias musculares en general. A continuación se presenta un estudio de reevaluación clínica y diagnóstico molecular de distrofinopatías en Costa Rica. Se encontraron deleciones en el gen de la distrofina, que podrían ser responsables del padecimiento en algunos pacientes y, por lo tanto, parecen confirmar el diagnóstico clínico. Así se ha podido identificar a varias mujeres como posibles portadoras de estas mutaciones, lo que puede incrementar el nivel de certeza para ofrecerles el consejo genético.
]]>
Los pacientes:
Treinta y un pacientes varones referidos al Hospital Nacional de Niños (HNN), diagnosticados inicialmente con distrofia muscular, cuyo cuadro clínico sugería que podría tratarse de distrofinopatías, fueron reevaluados en el Servicio de Genética y Metabolismo de este hospital. Veintitrés mostraron sintomatología propia de DMD y dos de BMD, mientras que seis no mostraron síntomas muy claros de alguna distrofinopatía. A estos pacientes se les tomó una muestra de sangre para los estudios de ADN. Además se realizó un diagnóstico prenatal a una portadora obligada (dos hermanos suyos ya han muerto por causa de DMD y su hijo mayor también padece la enfermedad) proveniente de otro país centroamericano. La muestra del producto en gestación se obtuvo por punción umbilical, mientras que la señora trajo consigo sangre de su hijo mayor. En todos los casos se contó con el consentimiento informado de los participantes o sus representantes legales.
Análisis molecular:
Las muestras de sangre se obtuvieron con vacutainers provistos de EDTA como anticoagulante. Los ADNs se extrajeron, con el método de sal (36) y se analizaron en el Instituto de Investigaciones en Salud (INISA) de la Universidad de Costa Rica.
Mediante PCR-multiplex se amplificaron fragmentos de 28 exones y de 2 promotores, en 4 reacciones independientes, a cada una de las muestras. Los perfiles de amplificación son los de Chamberlain et al. (33, 34) y Beggs et al. (35). Con uno de los lotes de imprimadores se hizo necesario disminuir la temperatura de hibridación de 600 a 570. Otra variante introducida fue el uso del buffer que acompaña a la Taq-polimerasa en lugar del buffer de Chamberlain et al (33).
Los productos de amplificación se separaron por electroforesis en geles de poliacrilamida de 16 cm de longitud y 1.5 mm de espesor (3% gel empacador de 2 cm y 10% gel separador de 12 cm). El voltaje aplicado fue de 120V constante durante 4-5 horas. Como amortiguador se empleó TBE 1X. Los geles fueron teñidos con nitrato de plata. La ausencia de bandas de amplificado se interpretó como evidencia de deleción del fragmento correspondiente.
Resultados
En ninguno de los pacientes con diagnóstico clínico dudoso de distrofinopatía se observaron deleciones. Este resultado permite sugerir con mayor grado de confianza que ellos padecen de otro tipo de distrofia muscular, por lo que no se tomaron en cuenta en lo concerniente a lo que sobre las distrofinopatías permite concluir este trabajo. No obstante, se puede demostrar el valor agregado de una prueba molecular en el diagnóstico y en sus consecuencias en el tratamiento y en el consejo genético. ]]>
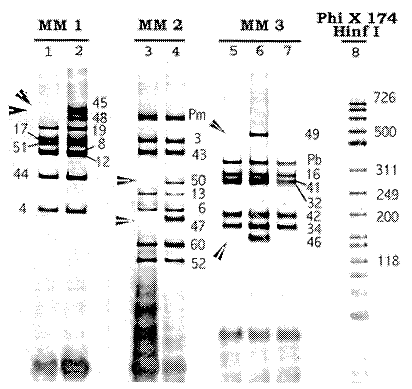
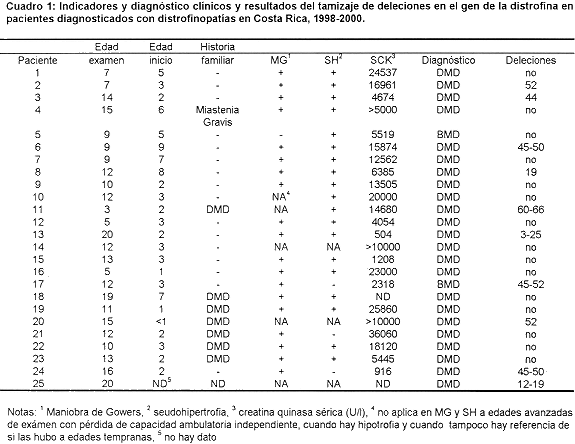
El resumen de los principales criterios diagnósticos, el diagnóstico y los resultados de¡ estudio molecular de los pacientes considerados como distrofinopáticos se presentan en el Cuadro 1. Dos pacientes, el 5 y el 17 se consideraron afectados por BMD, mientras que los otros 23 padecen de DMD. El tamizaje de deleciones se basa en la presencia o ausencia de los productos de amplificación de las distintas regiones génicas probadas, La Figura 1 muestra un gel con los productos de amplificación, con tresDe los 25 pacientes considerados distrofinopáticos, 10 presentaron deleciones. Seis mostraron deleciones que abarcan múltiples exones: en el paciente 13 la deleción cubre al menos 23 exones, del 23 al 25; en dos pacientes abarca al menos ocho exones en el 17 del 45 al 52 y en el 25 del 12 al 19; en el pacientell siete exones, del 60 al 66; en los pacientes 6 y 24 seis exones (del 45 al 50 en ambos). En cuatro pacientes la deleción comprende un único exón: en dos de ellos, el 2 y el 20 el exón 52; en el paciente 8 el exón 19 y en el paciente 3 el exón 44. Los otros 15 pacientes no presentaron deleciones. En vista de que los pacientes 18 y 19, así como los 21, 22 y 23 son hermanos, podemos decir que sólo el 45.5% (10 de 22) de las mutaciones estudiadas en esta muestra, contra 66.7% esperado, son deleciones. No obstante, la mayor parte de ellas (90%) se encuentran en las 2 regiones génicas de alta incidencia de deleciones, 30% alrededor de los exones 3-19 (esperado 20%) y 60% en torno a la región de los exones 44-52 (esperado 80%)
El diagnóstico prenatal practicado a solicitud de una portadora obligada, mostró que el producto en gestación, un varón según se determinó por ultrasonido, no portaba ninguna deleción, mientras que el paciente índice de la familia, quien está muy afectado, según referencia médica, tiene una enorme deleción que abarca al menos 37 exones, del 6 al 42. En una comunicación con la señora, meses después de que diera a luz, se informó que el niño había nacido bien y que su desarrollo era muy diferente en comparación con el mostrado por su hermano mayor afectado a su misma edad.
Discusión
El conocimiento de la estructura y función de muchos de los genes responsables de enfermedades genéticas, gracias al desarrollo de la biología molecular, ha permitido complementar y refinar el diagnóstico clínico, ya sea por el análisis directo del gen, a nivel de ADN, o por la evaluación de sus productos génicos, si se conocen. Así, se puede determinar, con un grado de certeza hasta hace poco insospechado, si alguno de ellos está involucrado en la patología observada.
La evolución del perfil de salud de Costa Rica muestra que, desde hace algunas décadas, las enfermedades crónicas han cobrado una mayor importancia relativa (37, 38). Sin duda el componente genético en la morbilidad de estos padecimientos es alto, por lo tanto es clara la necesidad de implementar métodos diagnósticos con base molecular. En el caso concreto de las distrofias musculares, el diagnóstico primario en Costa Rica es muy general y rara vez especifica el tipo de distrofia,, probablemente debido al traslape de sintomatologías observado entre algunas de ellas, a la falta de apoyo al clínico en pruebas de laboratorio que permitan el diagnóstico diferencial y a la deficiencia curricular en genética médica de muchos médicos generales, neurólogos y pediatras.
]]>
Con este estudio, se ha iniciado en Costa Rica la aplicación de este nuevo enfoque para refinar el diagnóstico de las más importantes de las distrofias musculares, las distrofinopatías. Para tener una idea de sus dimensiones en el país, se puede intentar la siguiente estimación. Entre 1981 y 2000 se informó de 811 834 nacimientos de varones (39). Si la tasa de incidencia para DMD es de 1/3 500 nacidos vivos y la de BMD de 1/18 500, entonces en ese periodo debieron nacer 232 niños con DMD y 44 con BMD total con distrofinopatías 276. Se escogió un período de 20 años pues es lo máximo que se espera que viva un paciente con DMD, así, los nacidos antes de 1981 deben haber fallecido ya.
De los 25 pacientes analizados, 10 presentaron deleciones. Esto tiene una consecuencia directa sobre el consejo genético, ya que permite buscar la misma mutación encontrada en el paciente índice en aquellas parientes con riesgo de ser portadoras, por ejemplo, madres, hermanas y tías maternas. La detección de portadoras es posible con técnicas como la hibridación fluorescente in situ (FISH) (40) y la cuantificación de dosis génica en productos de PCR (41, 42, 43, 44). Si alguna de ellas es portadora, tendría una probabilidad del 50% de que alguno de sus hijos varones fuera afectado. Por otra parte, si no lo fuera, tiene un riesgo muy bajo, el correspondiente a la mitad de la tasa de mutación de este gen, que es el mismo de cualquier otra mujer de la población. En ambos casos es claro el efecto de tal diagnóstico en sus decisiones sobre su futuro reproductivo, lo que sin duda traería consigo un alivio emocional a muchas familias y al sistema de salud.
Figura 1: Electroforesis en gel de poliacrilamida (10%) mostrando productos de amplificación de PCR-múltiplex con 3 combinaciones de imprimadores (MM1, MM2 y MM3) en el gen de la dístrofina y visualizados con nitrato de plata. Los carriles 1, 3, 5 y 7 corresponden a la muestra del paciente 6, mientras que los carriles 2, 4 y 6 son de un control normal. El carril 8 es del marcador de tamaño de las bandas, las que se indican, en pares de bases, con números a la derecha de algunas de ellas. Las flechas señalan la ausencia de bandas (falta de amplificación) en las respectivas M M del paciente, bandas que si están presentes en el control. Los números al lado de las bandas corresponden al número del exón o promotor (Pm o Pb) amplificado. La ausencia de amplificación se atribuye a la pérdida (deleción) del segmento correspondiente en el gen. El paciente muestra falta de amplificación de los exones 46 y 48 en el MM1 (carril 1), de los exofies 47 y 50 en el MM2 (carril 3) y de los exones 46 y 49 en el MM3 (carriles 5 y 7). Nótese que los exones 44 y 51 si amplificaron en la muestra del paciente (carril 1) lo que permite demarcar la extensión de la delecíón entre los exones 45 y 50.
Otro pequeño ejercicio teórico podría ayudar a estimar el impacto que tendrían las distrofinopatías sin medidas de prevención apoyadas en un consejo genético basado en análisis moleculares. Según proyecciones del Observatorio Demográfico del Programa Centroamericano de Población, de la Universidad de Costa Rica (45), a finales del año 2000 habría en Costa Rica 1 567 094 mujeres entre los 0 y los 44 años de edad. Si la tasa de mutación () compensa a la tasa de eliminación de alelos por muerte o incapacidad reproductiva de los varones distrofinopáticos (coeficiente de selección s), sea = s (46, 47), la proporción de portadoras sería: ]]>
2pq para DMD, dondeDe esta manera, habría 896 portadoras de DMD y 172 de BMD (total 1068). Si cada mujer tiene en promedio 2.4 hijos, estas portadoras habrán procreado al año 2044, cuando se espera que las que ahora tiene 0 años han concluido su ciclo reproductivo, un total de 2563 descendientes, 641 de ellos varones afectados con distrofinopatías y un número igual de hijas portadoras. Algunos de estos descendientes han nacido ya, por ejemplo, los hijos de la cohorte que tiene 45 años ya nacieron; de hecho, los pacientes analizados aquí son de ese grupo y algunos de esos varones afectados ya murieron. Aparte del impacto emocional para las familias con niños distrofinopáticos, el costo de estos pacientes para el sistema de salud sería muy elevado en vista del mayor número de hospitalizaciones, de las estancias más prolongadas y del mayor número de intervenciones quirúrgicas que los pacientes con enfermedades genéticas requieren (48).
En cuanto a los pacientes en quienes no se encontraron deleciones, no se puede concluir que no son distrofinopáticos, ya que la técnica utilizada detecta únicamente deleciones. Debe recordarse que un tercio de las mutaciones responsables de las distrofinopatías son mutaciones de punto (49) y duplicaciones (25). Tales alteraciones, ocurren aleatoriamente a lo largo del gen, y dadas las dimensiones de este, no se ha podido desarrollar una estrategia racional de tamizaje molecular, por lo que no se puede avanzar mucho al respecto en asuntos de prevención con la metodología implementada para este estudio.
No obstante, con la implementación de esta metodología se puede iniciar el estudio de la epidemiología de estas enfermedades en Costa Rica. Al momento, la proporción de pacientes con deleciones (10122 = 45.5%) está por debajo de lo esperado (213 = 66.66%) según investigaciones en otras partes del mundo, pero la muestra no es suficiente para ser considerada válida, además de que representa, principalmente, una parte de la casuística del Valle Central, la que es referida al H ospital Nacional de Niños. El objetivo podría cumplirse con la colaboración de la comunidad médica nacional, si se comunicaran, al Servicio de Genética y Metabolismo del Hospital Nacional de Niños, los casos posibles candidatos a ser afectados con distrofinopatías. ]]>
Conclusiones
El diagnóstico diferencial de las distintas distrofias musculares se ve dificultado por el traslape de síntomas y la falta de experiencia de los clínicos con enfermedades genéticas. Esto evidencia la necesidad de implementar métodos diagnósticos más discriminantes en Costa Rica. El conocimiento de las causas proximales de muchas enfermedades genéticas permite implementar métodos diagnósticos a nivel del ADN. La utilidad de estos métodos se ha demostrado en este trabajo con el caso de las distrofinopatías. La identificación de pacientes con deleciones permite además buscar estas mutaciones en las mujeres parientes del paciente que tendrían un riesgo mayor de ser portadoras de la mutación y así ofrecerles consejo genético con mejor base. Además, la etodología podría servir para determinar la epidemiología de estos padecimientos.
Agradecimientos
A los señores Jorge Monge y a Víctor Castillo por su ayuda técnica y a los Dres. Kay Sanders Mangel por efectuar la toma de muestra de cordón umbilical y Mario Barrantes González por referirnos a un paciente con DMD. Este trabajo fue financiado por la Vicerrectoría de Investigación de la Universidad de Costa Rica, proyecto número 742-97-253.
Referencias
1. Emery AEH. Duchenne Muscular Dystrophy. Oxford University Press, Great Britain. 1993; 391 p. [ Links ]
2.Speer A, Oexle K. Muskeldystrophien. En: Ganten D, Ruckpaul K eds. Monogen bedingte Erbkrankheiten. Teil 1. Springer-Verlag, Berlin. 1999; 3-30. [ Links ]
3. Emery AEH. Population frequencies of inherited neuromuscular diseases. A world survey. Neuromusc Disord 1991; 1: 19-29. [ Links ]
4.Moser H, Emery AEH. The manifesting carrier in Duchenne muscular Dystrophy. Clin Genet 1974; 5: 271-284. [ Links ]
5. Emery AEH. Muscle histology and creatinine kinase levels in the foetus in Duchenne muscular dystrophy. Nature 1977; 266: 472-473. [ Links ]
6. Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet 1984; 66:17-40. [ Links ]
7. Becker PE, Kiener F. Eine neue Xchomosomale Muskeidystrophie. Arch Psychiatr Nervenkrankheiten 1955; 193:427-448. [ Links ]
8. Murray JM, Davies KE, Harper PS, Meredith L, Mueller CR, Williamson R. Linkage relationship of a cloned DNA sequence on the short arm of the X chromosome to Duchenne muscular dystrophy. Nature 1982; 300:69-71. [ Links ]
9. Kunkel LM, Monaco AP, Middlesworth W, Ochs HD, Latt SA. Specific cloning of DNA fragments absent from the DNA of a male patient with an X chromosome deletion. Proc Natl Acad Sci USA 1985; 82: 4778-4782. [ Links ]
10. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) CDNA and preliminary genomic organization of the DMD gene in normal and affected individuas. Cell 1987; 50: 509-517. [ Links ]
11. Roberts RG, Coffey AJ, Bobrow M, Bentley DR. Exon structure of the human dystrophy gene. Genomics 1993; 16:536-538 [ Links ]
12. Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nature Genet 1993; 3: 283-91. [ Links ]
13. Tinsley JM, Blake DJ, Pearce M. Knight AE, Kendrick-Jones J, Davies KE. Dystrophin and related proteins. Curr Op Genet Devel 1993; 3: 484-90. [ Links ]
14. Pillers DAM, Bulman DE, Weleber RG, et al. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nature Genet 1993; 4:82-86 [ Links ]
15. Campbell K, Kahl S. Association of dystrophin and an integral membrane glycoprotein. Nature 1989; 338:259-262. [ Links ]
16. Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991; 66: 1121. [ Links ]
17. Muntoni F, Wilson L, Marrosu G et al. A mutation in the dystrophin gene selectively affecting dystrophin expression in the hearth. J Clin lnvest 1995;96:693-699. [ Links ]
18. Darras B, Blattner P, Harper J, Spiro A, Alter S, Francke U. lntragenic deletions in 21 Duchenne muscular dystrophy (DMD)/Becker muscular dystrophy (BMD) families studied with the dystrophin cDNA: location of breakpoints on Hindlll and Bglll exoncontaining fragments maps, meiotic and mitotic origin of the mutations. Am J Hum Genet 1988; 43: 620-629. [ Links ]
19. Gillard EF, Chamberlain, JS, Murphy EG, et al. Molecular and phenotypic analysis of patients with deletions within the deletion-rich region of the Duchenne muscular dystrophy (DMD) gene. Am J Hum Genet 1989; 45: 507-520.
20. Coral-Vazquez R, Arenas D, Cisneros B, et al. Pattern of deletions of the dystrophin gene in Mexican Duchenne/Becker Muscular Dystrophy patients. The use of new designed primers for the analysis of the major deletion "hot spot" region. Am J Med Genet 1997; 70: 240-246. [ Links ]
21. Florentin L, Mavrou A, Kekou K, Metaxotou C. Deletion patterns of Duchenne and Becker muscular dystrophies in Greece. J Med Genet 1995; 32: 48-51. [ Links ]
22. Peterlin B, Zidar J, Meznaric-Petrusa M, Zupancic N. Genetic epidemiology of Duchenne and Becker muscular dystrophy in Siovenia. Clin Genet 1997; 51: 94-97. [ Links ]
23. Banerjee M, Verma IC. Are there ethnic differences in deletions in the dystrophin gene? Am J Med Genet 1997; 68: 152-157. [ Links ]
24. Hiraishi Y, Kato S, Ishihara T, Takano T. Quantitative Southern biot analysis of the dystrophin gene of Japanese patients with Duchenne or Becker muscular dystrophy: a high frequency of duplications. J Med Genet 1992; 29:897-901. [ Links ]
25. u X, Ray P, Murphy G, Thompson M, Worton R. Duplicational mutation at the Duchenne muscular dystrophy locus: its frequency, distribution, origin, and phenotype-genotype correlation. Am J Hum Genet 1990; 46: 682695. [ Links ]
26. Tuffery S, Bareil C, Demaille J, Claustres M. Four novel dystrophin point mutations: Detection by protein truncation test and transcript analysis in lymphocytes from Duchenne Muscular Dystrophy patients. Eur J Hum Genet 1996; 4: 143-152. [ Links ]
27. Baxter PS, Maitby EL, Quarrell O. Xp21 Muscular dystrophy due to X chromosome inversion. Neurology 1997; 49: 260. [ Links ]
28. Oudet C, Heilig R, Hanauer A, Mandel JL. Nonradioactive assay for new microsatellite polymorphisms at the 5' end of the dystrophin gene, and estimation of intragenic recombination. Am J Hum Genet 1991; 49: 311-319. [ Links ]
29. Oudet C, Hanauer A, Clemens P, Caskey T, Mandel JL. Two hot spots of recombination in the DMD gene correlate with the-deletion prone regions. Human Molecular Genetics 1992; 8: 599-603. [ Links ]
30. McNaughton JC, Hughes G, Jones WA, Stockwell PA, Kiamut HJ, Petersen GB. The evolution of an intron: Analysis of a long, deletion-prone intron in the human dystrophin gene. Genomics 1997; 40: 294-304. [ Links ]
31. Pizzuti A, Piertte M, Fenwick RG, Gibbs RA, Caskey CT. A Transposon-like element in the deletion-prone region of the dystrophin gene. Genomics 1992; 13: 594-600. [ Links ]
32. Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985; 230:1350-1354. [ Links ]
33. Chamberlain JS, Gibbs RA, Ranier JE. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucl Acids Res 1988; 16: 11141. [ Links ]
34. Chamberlain JS, Gibss RA, Ranier JE, Caskey CT. Multiplex PCR for the diagnosis of Duchenne muscular dystrophy. En: lnnis MA, Gelfand DH, Sninsky JJ, White TS, eds. PCR Protocols: A Guide to Methods and Applications. Academic Press, lnc. 1990; 272-281. [ Links ]
35. Beggs A, Koenig M, Boyce F, Kunkel L. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum
Genet 1990; 86: 45-48.
36. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 1988; 16:1215. [ Links ]
37. Mohs E. Infectious diseases and health in Costa Rica- the development of a new paradigm. Pediat Infect Dis 1982; 1:212-216. [ Links ]
38. Sáenz R, Gomez X. Análisis de la situación de salud con base en las condiciones de vida. Costa Rica 1970-1996. Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud (INCIENSA), Cartago, Ministerio de Salud, Costa Rica. 1998; 91 pp. [ Links ]
39. Programa Centroamericano de Población, Universidad de Costa Rica. [ Links ]
40. Tocharoentanaphol Ch, Cremer M, Schröck E, et al. Multicolor fluorescence in situ hybridization on metaphase chromosomes and interphase Halo-preparations using cosmid and YAC clones for the simultaneous high resolution mapping of deletions in the dystrophin gene. Hum Genet 1994; 93: 229- 35. [ Links ]
41. Schwartz LS, Tarleton J, Popovich B, Seitzer WK, Hoffman EP. Fluorescent multiplex linkage analysis and carrier detection for Duchenne/Becker Muscular Dystrophy. Am J Hum Genet 1992; 51: 721-729. [ Links ]
42. Mansfield ES, Robertson JM, Lebo RV, et al. Duchenne/Becker Muscular Dystrophy carrier detection using quantitative PCR and fluorescence-based strategies. Am J Med Genet 1993; 48: 200-208. [ Links ]
43. Yau SC, Bobrow M, Mathew CG, Abss SJ. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet 1996; 33: 550-9.
44. Fortina P, Cheng J, Shoffner M, et al. Diagnosis of the Duchenne/Becker muscular dystrophy and quantitative identification of carrier status by use of entangled solution capillary electrophoresis. Clin Chem 1997; 43: 745-751. [ Links ]
45. Haldane JBS. The Causes of Evolution Longmans, Green. London 1932. [ Links ]
46. Haldane JBS. The rate of spontaneous mutation of a human gene. J Genet 1935 31:317-326 [ Links ]
47. de Céspedes C, Umaña L, Yock 1, Bustamante M, Atkins AT. Frecuencia y demanda de atención médica de las enfermedades genéticas en el Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera". Acta Ped Cost 1996; 10:53-60.
48. Yau SC, Bobrow M, Roberts RG, Mathew CG. Direct diagnosis of carriers of point mutations in Duchenne muscular dystrophy. Lancet 1993; 341: 273-75. [ Links ]
1Bióloga, genetista, Escuela de Biología, Universidad de Costa Rica, Instituto de Investigaciones en Salud (INISA).
2Pediatra, Especialista en Genética Servicio de Genética y Metabolismo, Hospital Nacional de Niños Médico. ]]>
3Bioquímico, Escuela de Medicina, UCR4Biólogo, genetista, Dr. Instituto de Investigaciones en Salud (INISA)
Abstract
Objective
To introduce molecular-genetic methods to the study of dystrophinopathies in Costa Rica.
Materials and methods
Thirty-one male patients diagnosed with muscular dystrophy, which could be affected with a dystrophinopathy, were clinically re-examined. Twenty-three had DMD, and two BMD and six showed no clear-cut symptoms of a dystrophinopathy. DNA samples were screened by multiplex PCR for deletions in the dystrophin gene. A prenatal diagnosis was made upon request from an obligate carrier.
Results
Ten patients showed deletions: one has a deletion of 23 exons (3-25); two have deletions of eight exons (one 45-52, the other 12-19); another has a deletion of 7 exons (60-66); two have a deletion of 6 exons (both 45-50). The four remaining patients showed single-exon deletions: exon 52 (two patients), exon 19 and exon 44. The older son of the obligate carrier, affected with DMD, showed a deletion of 36 exons (6-42), whereas the fetus has no deletion. None of the patients with unclear diagnosis had deletions. ]]>
ConclusionThe overiap of symptoms observed among different muscular dystrophies, the lack of available laboratory tests and the scarcity of physicians trained in Medical Genetics, evidence the need to implement more discriminatory diagnostic methods in Costa Rica. The identification of patients with deletions allows the search for the same mutations in women at risk of being carriers and to offer them a more accurate genetic counseling. ]]>