Objetivo
Estimar la dosis génica en el gen de la distrofina a mujeres a riesgo de ser portadoras de deleciones.
Sitio de realización
INISA y Escuela de Biología, Universidad de Costa Rica. ]]>
Materiales y métodosSe obtuvo ADN de 15 mujeres, emparentadas por línea materna con pacientes afectados con DMD o BMD causadas por deleciones en el gen de la distrofina, y que por lo tanto podrían ser portadoras de esas mutaciones, para amplificar de 7 a 9 regiones del gen por medio de PCR-múltiplex. Para estimar la dosis génica, se marcó uno de los imprimadores (el F) de cada par con el fluorocromo 6-FAM; los productos de amplificación se separaron y se cuantificaron por electroforesis capilar fluorescente.
Resultados
Todas las posibles portadoras mostraron dosis génicas como las de mujeres sin deleciones.
Conclusiones
El resultado indica que todos los pacientes son producto de mutaciones de novo, lo que es inesperado, pues datos de otros países muestran que ese es el caso de a sólo 113 de los pacientes. Además, la evaluación ofrece mejores elementos de juicio para brindar consejo genético.
Palabras clave: Distrofina, Duchenne, Becker-Kiener, portadoras, deleciones, herencia ligada al cromosoma X, PCR, electroforesis capilar fluorescente, dosis génica.
]]>
La DMD es la más frecuente con una incidencia de 1:3500 niños varones nacidos vivos (4) y la más severa de ambas formas de distrofinopatía, terminando su progresión con la muerte de los pacientes alrededor de los 20 años de edad (5). La BMD, con una incidencia de 1:18500 (4) es menos severa; algunos de los pacientes llegan incluso a reproducirse (7).
La mayor parte, dos tercios, de las mutaciones que causan las distrofinopatías son deleciones, o pérdidas de material genético del gen (12). Del 5 al 10% de los casos (13), con un notable 14% en Japón (14), son duplicaciones. El resto se deben a mutaciones de punto(15) e inversiones (16).
El conocimiento de la estructura génica y del patrón de mutaciones ha permitido desarrollar técnicas moleculares para detectar la mayor parte de las mutaciones utilizando PCR-múltiplex (17, 18), una variante de la PCR que permite amplificar, en una única reacción, hasta nueve segmentos del gen de la distrofina. Como la distribución de las deleciones no es aleatoria, sino que se concentra alrededor de 2 regiones, la utilización de los dos combinaciones de imprimadores permite detectar más del 98% de las deleciones (18). Esta metodología se utilizó para buscar deleciones en pacientes aparentemente distrofinopáticos en Costa Rica (19).
Una variante de la PCR-múltiplex se puede usar para evaluar si mujeres que, por ser parientes en primer grado por parte materna de pacientes con deleciones, podrían ser portadoras de esas mismas mutaciones y así ser madres potenciales de niños afectados con estas graves enfermedades. La mutación que los pacientes reciben de sus madres puede haberse heredado porque ella es portadora o porque la mutación se ha producido de novo, esto es, en la línea germinal de la madre. Según sea el caso, las consecuencias para el consejo genético son diversas. Vale recordar que por probabilidades, la mitad de los hijos varones de las mujer portadoras de una mutación en un rasgo ligado al cromosoma X padecen de la enfermedad, y la mitad de sus hijas serán portadoras al igual que ellas, por el contrario, si las mujeres no son portadoras tienen un riesgo equivalente a la incidencia de la enfermedad en la población, lo que señala la importancia de determinar el status de estas mujeres para efectos de consejo genético.
La variante de la técnica consiste en marcar los productos de amplificación por PCR con una molécula fluorescente, lo que permite cuantificar el número de moléculas amplificadas según sea la intensidad de la señal durante la electroforesis capilar. Dos moléculas rendirán el doble de señal fluorescente que una molécula. De esta manera, una mujer normal tendría dos copias completas del gen de la distrofina y cada copia sirve de base para que, por medio de la PCR, se amplifiquen moléculas marcadas. Por el contrario, una mujer con deleción en uno de los dos genes de la distrofina tiene sólo una copia completa del gen, por lo que sólo tendrá la mitad de las moléculas, con respecto a una mujer normal, sirviendo como base para la amplificación de moléculas marcadas. Así, a partir de esa única copia, se produce la mitad de moléculas marcadas que produce una mujer normal en un proceso similar. Entonces, el número de moléculas marcadas producidas por medio de PCR es un reflejo de la dosis génica que tiene la mujer e indica si ella, la posible portadora, acarrea la misma deleción encontrada en el paciente índice de su familia.
En este trabajo se estimaron las dosis génicas a mujeres emparentadas en primer grado, por vía materna, con pacientes con
deleciones en el gen de la distrofina, lo que les produce DMD o BMD (1 g), con el fin detener más y mejores elementos de juicio para brindarles consejo genético y para estimar el impacto de las mutaciones de novo en la incidencia de las deleciones en este gen en Costa Rica.
Materiales y Métodos ]]>
Los sujetos de estudio.En un estudio anterior (19) se logró determinar que algunos pacientes con DMD o BMD sufren la enfermedad por causa de deleciones en el gen de la distrofina. Esto permitió identificar a 15 mujeres, parientes en primer grado por vía materna de 6 de estos pacientes, que tienen por lo tanto altas posibilidades de ser portadoras de las mutaciones que afectan a los pacientes índice de sus respectivas familias. Ellas son la madre, 8a,y tres hermanas, 8b, 8c y 8d, del paciente 8 (se usará el número del paciente designado en Sancho et al.(19), quien tiene una deleción del exón 19; la madre, 3a, y tres hermanas 3b, 3c y 3d, del paciente 3, quien tiene una deleción del exón 44; la madre, 13a, del paciente 13, que tiene una deleción que abarca del exón 3 al 25; la madre, 6a, y una hermana, 6b del paciente 6, afectado con una deleción que involucro a la región de los exones 45 al 50; la madre, 17a, la tía, 17b, y la abuela materna, 17c, del paciente 17, con deleción de los exones 45 al 52; y la madre, 24a, del paciente 24, con deleción de los exones 45 al 50. En ninguno de los casos hay antecedentes de distrofinopatías.
Muestras de ADN de estas mujeres se extrajeron de sangre total (Vacutainers con EDTA) usando el método de sal (20) y se usaron para cuantificar sus dosis génicas amplificando diversas regiones del gen de la distrofina mediante PCR-múltiplex, usando los imprimadores y el principio diseñados por Chamberlain et al. (17), y separando y cuantificando los productos de amplificación
mediante electroforesis capilar fluorescente (21).
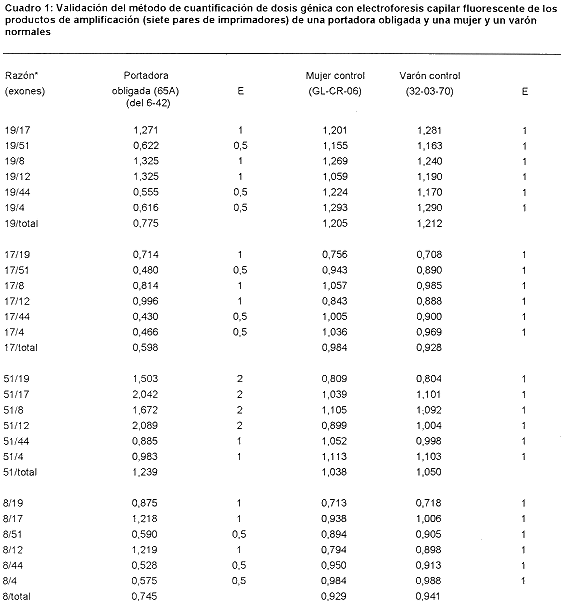
Los valores de referencia normales se obtuvieron determinando la dosis génica a 12 mujeres independientes, sin antecedentes de distrofinopatías en sus familias. Además, se probó la sensibilidad del método estimando la dosis génica de una portadora obligada de una deleción (dos hermanos suyos ya fallecieron víctimas DMD y su hijo mayor está también afectado y tiene una deleción de 37 exones, del 6 al 42 (19), y de dos controles normales, una señora y un hombre. También se hicieron amplificaciones de los pacientes índice y del hijo de la portadora obligada como controles negativos de la amplificación de sus exones deletados.
Las muestras de todas las personas se obtuvieron con su consentimiento informado o el de sus representantes legales.
Reacciones de amplificación por PCR-múltiplex.
Las amplificaciones se hicieron utilizando como base la combinación de 9 pares imprimadores (exones 4, 8, 12, 17, 19, 44, 45, 48 y 51) y el perfil de PCR de Chamberlain et al. (17) con los ajustes apuntados por Sancho et al. (19). Dos modificaciones adicionales se hacen necesarias para poder evaluar la dosis génica con electroforesis fluorescente. La primera es la utilización de uno de los imprimadores, (el F) del par correspondiente a cada exón a amplificar, marcado con un fluorocromo, en este caso 6 carboxifluoresceína o 6-FAM, en el extremo 5' de la molécula. Esta modificación permite tener una única señal fluorescente por molécula amplificada. La segunda modificación fue la de reducir el número de ciclos de amplificación a 18, lo que hace que el número de moléculas obtenidas a ese punto sea proporcional al número de moléculas iniciales, sean, las correspondientes al número de regiones del gen, o dosis génica, al encontrarse la PCR en su fase logarítmica ya que amplificaciones de más de 20 ciclos no guardan esta relación (21). Así, las portadoras de deleciones amplificarán la mitad de las moléculas en la región a probar, dosis génica simple, con respecto a las mujeres con dos genes normales, completos, dosis génicas dobles.
Los controles de sensibilidad de la prueba y las parientes de los pacientes 8, 3 y 13 se amplificaron con 7 pares de imprimadores reduciendo así el nivel de Complejidad de la reacción. Se dejaron por fuera a los exones 45 y 48 que en estas personas eran innecesarios para efectuar la evaluación. Las parientes de los pacientes 6, 17 y 24 se amplificaron con los 9 pares de imprimadores, pues el exón 45 era de los que había que probar como posible deletado en esas familias.
Electroforesis capilar ]]>
El análisis mediante electroforesis capilar se hizo con un Analizador Genético ABI 310 (Applied Biosystems) utilizando el programa ABI PRISM 310 Genetic Analyzer Data Collection, versión 1.0.4 de la Corporación Perkin Elmer (1997), para la recolección de lo datos. El proceso fue gobernado con un computador Power PC de Macintosh equipado con un procesador G3.Los productos de amplificación se diluyeron para las electroforesis capilares así: amplificado 3µl; marcador de tamaño molecular (ROX 500 o ROX 1000) 0.5µl; formamida desionizada 10.5µl. Las diluciones se desnaturalizaron por 5 minutos a 95°C, pasándose la muestra de inmediato a hielo. La separación de fragmentos se hizo en capilares de 47 cm x 50µm (No. 402839) utilizando el polímero "Performance Optimized Polymer 4 (POP4)" (No. 402838). Como solución amortiguadora se usó "310 Genetic Analyzer Buffer con EDTA" (No. 402824) diluida a 1X. El módulo utilizado fue GS STR POP (1 ml) con 5 segundos de inyección a 15 kV. Las corridas fueron a 15kV por tiempos de entre 25 y 45 minutos. La matriz empleada fue la "Matrix Filter D" que se incluye en el programa como parte de su grupo básico.
Determinación de la dosis génica en candidatas a ser portadoras de deleciones
Para determinar la dosis génica se usaron los valores de las áreas de las señales correspondientes a los amplificados de cada exón. El procedimiento consistió en obtener razones dividiendo el valor del área de cada exón entre los valores de las áreas de los demás exones. De esta manera se reducen los errores de variación entre diferentes reacciones de amplificación. Los valores de las razones obtenidas en las posibles portadoras de deleciones se dividieron entre las razones correspondientes a los promedios de las 12 mujeres control. Si la mujer no es portadora de deleciones, sus razones han de ser equivalentes a las del promedio de las mujeres control, obteniéndose por lo tanto valores de 1. Si por el contrario, la mujer es portadora de una deleción, los valores de sus exones deletados con respecto al promedio de las mujeres normales serán de 0.5 cuando el exón deletado sea el numerador, de 1.0 cuando la deleción involucro a varios exones, por lo que ambos estarían en dosis simples y las razones entre ellos son equivalentes a las dosis dobles en las mujeres normales, o de 2.0 cuando el exón deletado sea el denominador. Estos valores se calcularon con el programa Excel 2000 (Microsoft, 1999)
Resultados
Validación del método
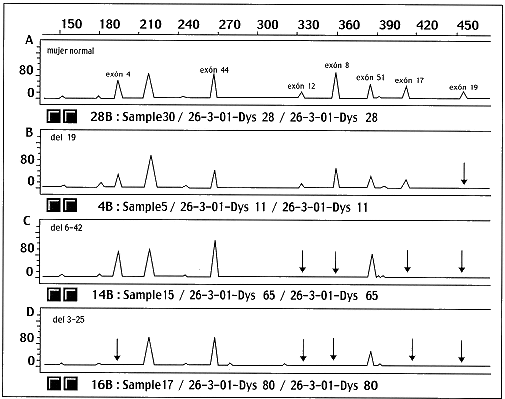 Figura 1: Electroferogramas de los productos de amplificación de PCR-múltiplex usando 7 pares de imprimadores de una mujer control y tres pacientes afectados con diversas deleciones. Las flechas indican la ausencia de señales de aquellos exones que no amplificaron y por lo tanto se encuentran deletados. El panel lA corresponde a una mujer normal donde aparecen los picos (señales) de los siete exones que amplifican para este grupo de imprimadores. El panel lB pertenece a un paciente que presenta deleción del exón 19, por lo que, está ausente el pico correspondiente a este exón. El panel 1C es de un paciente con una deleción que abarca los exones 6-42, por lo que no aparecen los picos de los exones 8, 12, 17 y 19. El panel 1D pertenece a un paciente afectado con la deleción de los exones 3-25, según se muestra, están ausentes los exones 4, 12, 8, 17 y 19. El área bajo los picos correspondientes a cada región amplificada es lo que sirve para cuantificar la dosis génica de las posibles portadoras.
Figura 1: Electroferogramas de los productos de amplificación de PCR-múltiplex usando 7 pares de imprimadores de una mujer control y tres pacientes afectados con diversas deleciones. Las flechas indican la ausencia de señales de aquellos exones que no amplificaron y por lo tanto se encuentran deletados. El panel lA corresponde a una mujer normal donde aparecen los picos (señales) de los siete exones que amplifican para este grupo de imprimadores. El panel lB pertenece a un paciente que presenta deleción del exón 19, por lo que, está ausente el pico correspondiente a este exón. El panel 1C es de un paciente con una deleción que abarca los exones 6-42, por lo que no aparecen los picos de los exones 8, 12, 17 y 19. El panel 1D pertenece a un paciente afectado con la deleción de los exones 3-25, según se muestra, están ausentes los exones 4, 12, 8, 17 y 19. El área bajo los picos correspondientes a cada región amplificada es lo que sirve para cuantificar la dosis génica de las posibles portadoras.
B. Estimación de la dosis génica de una portadora obligada y de dos controles normales
Para comprobar la sensibilidad del método en la detección de diferencias de dosis génica, se evaluaron las muestras de una portadora obligada de una deleción de los exones 6 al 42 y de dos controles normales, una mujer y un varón.
El Cuadro 1 presenta las razones de las áreas de los distintos picos de estas muestras con respecto a las correspondientes a los promedios de las mujeres normales, las que se comparan los valores esperados (E). En el caso de la portadora obligada los valores coinciden con los esperados de acuerdo a la deleción presente en su hijo, mientras que los controles normales muestran valores equivalentes a los de los promedios de las mujeres normales, lo que muestra que el método es lo suficientemente confiable para la cuantificación de dosis génica
Posibles portadoras.
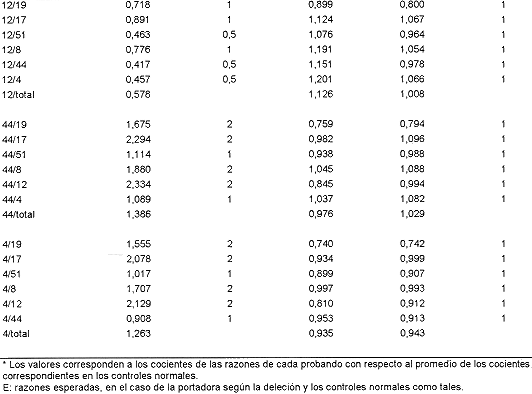
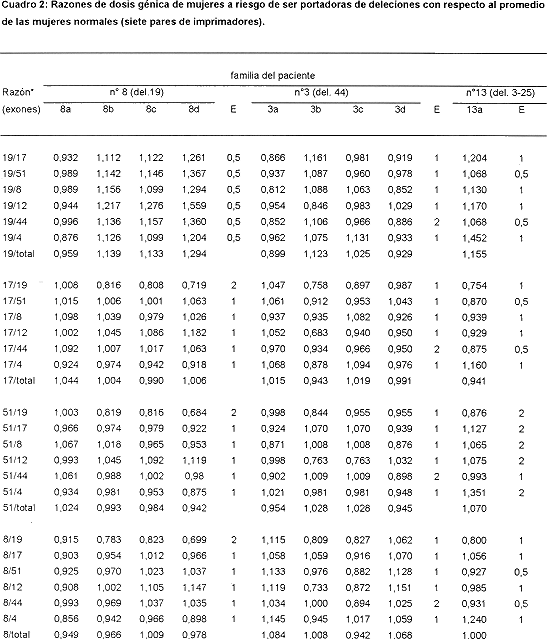
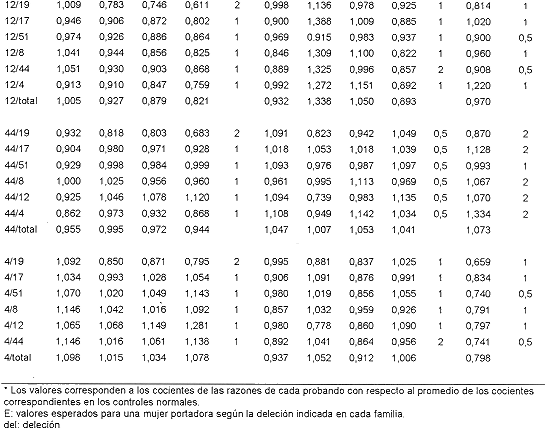
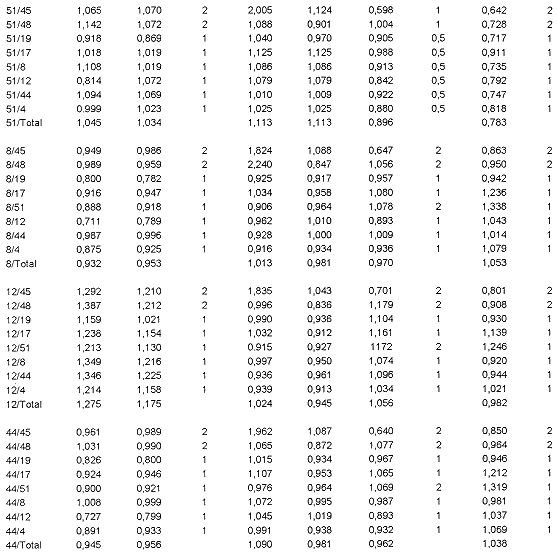
En el Cuadro 2 se muestran las razones obtenidas para aquellas posibles portadoras cuya dosis génica se estimó con el subgrupo de 7 pares de imprimadores. Para la familia del paciente n ° 8, se analizaron las razones obtenidas para su madre (8a) y sus tres hermanas (8b, 8c, 8d). Los valores para cada exón tienden a 1 con excepción de algunos valores que involucran al exón 19. Para este exón los valores son algo superiores a 1, cuando se encuentra como numerador e inferiores cuando está como denominador. Esta variación se observó para ese exón a lo largo de todo el estudio; de hecho, es de los que mayor desviación estándar tiene en la estimación de los promedios normales (datos no mostrados). Sin embargo, al considerarse en conjunto todas las razones obtenidas, se puede concluir que ni la madre ni las hermanas son portadoras de la deleción, pues las razones que con él se obtienen no se aproximan a los valores esperados en caso de que ellas lo fueran, todo lo contrario se alejan de ese valor hacia los esperados en mujeres normales. El Cuadro 2 muestra también las razones de las familiares del paciente 3, madre (3a) y hermanas (3b, 3c, y 3d), así como los de la madre (12a) del paciente 12. Las razones de todas son prácticamente de 1, o sea, las correspondientes a mujeres con dosis génica normal.
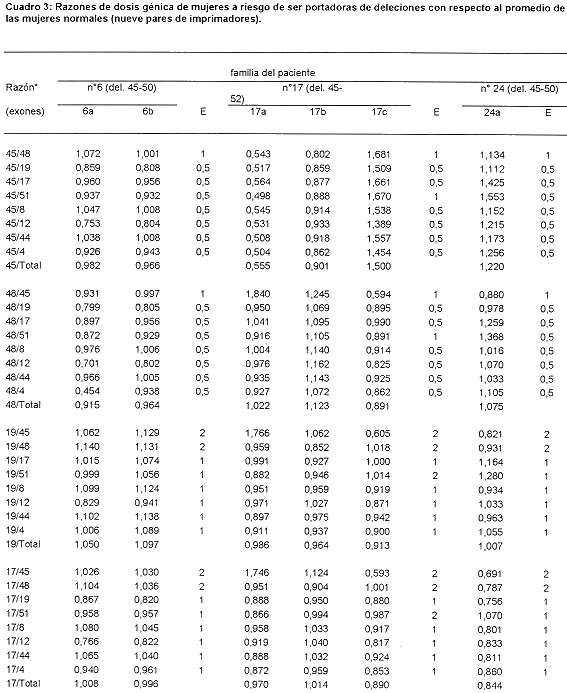
El Cuadro 3 presenta las razones obtenidas para aquellas posibles portadoras cuya dosis génica se estimó con los 9 pares de imprimadores; estas son, la madre, 6a, y la hermana, 6b, del paciente 6; la madre, 17a, una tía materna, 17b, y la abuela materna, 17c, del paciente 17, y la madre, 24a, del paciente 24. Las valoraciones indican que ninguna de ellas es portadora de la deleción encontrada en los, pacientes índice de sus familias. Merecen consideración aparte algunos de los valores obtenidos con las razones que involucran al exón 45 en la señora 17a, que podrían indicar que se trata de una portadora (45/19, 45/17, 45/8, 45/12, 45/44 y 45/4, 19/45 y 17/45). Esa posibilidad se descarta si se toma en cuenta que ella sería portadora de una deleción del exón 45 al 52, que involucraría a los exones 48 y 51 incluidos en la combinación de imprimadores empleada y que rinden valores de mujer con dosis génica normal. Además, los valores de alrededor de 0.5 se observan aún en aquellas relaciones en que se esperan valores de 1.0, lo que indica que esos valores se deben a algún artificio técnico.
Vistos en conjunto, los datos obtenidos indican que todos los pacientes índices parecen deber las deleciones que los afectan a mutaciones de novo, es decir ocurridas en la línea germinal de sus madres, resultado inesperado, dado que según antecedentes de otros países, estos son sólo 113 de los casos observados. ]]>
La disponibilidad de técnicas que permiten el análisis de los defectos genéticos al nivel del ADN tiene impacto directo en el
diagnóstico diferencial y en el consejo genético, lo que a su vez tendría consecuencias positivas en los sistemas de salud al poderse estimar el impacto epidemiológico de estos padecimientos y por lo tanto preparar al sistema para enfrentarlos racionalmente. Así, mientras la terapia génica siga siendo una promesa, la mejor forma de enfrentar estas enfermedades es la prevención; en este sentido, el consejo genético basado en pruebas más discriminantes para determinar el genotipo de las posibles personas portadoras de genes de predisposición aumentada a enfermedades, debería, en el mejor de los casos, contribuir a disminuir, primero, la carga emocional que pesa sobre las familias con pacientes afectados con enfermedades genéticas y, segundo, la carga social que esos casos implican (22).
El presente trabajo, que se deriva de un estudio previo (19) es un primer paso para implementar técnicas moleculares modernas, PCR-múltiplex (17), y electroforesis capilar fluorescente (21), en Costa Rica, con el fin de mejorar los criterios para ofrecer consejo genético a posibles portadoras de deleciones en el gen de la distrofina.
La metodología empleada resultó ser lo suficientemente específica, como lo demostraron los controles negativos y positivos de deleciones (los pacientes índice de cada familia analizada y los controles normales) y lo suficientemente sensible para la determinación de diferencias en la dosis génica, como lo demostraron la pruebas en las muestras de la portadora obligada de una extensa deleción y las mujeres normales. Los valores algo atípicos observados con los exones 19 y 45 son probablemente debidos al hecho de que el perfil de las reacciones de amplificación no es óptimo para todos los pares de imprimadores, esto es, no todos amplifican con la misma eficiencia pues sus temperaturas óptimas de hibridación y de polimerización difieren de un par a otro debido a sus diferentes secuencias. No obstante, la evaluación conjunta de todas las pruebas, tomando en cuenta todas las variables disponibles, como el conocimiento previo de la extensión de la deleción en el paciente índice, como se hizo en el caso de la señora 17a y el exón 45, o el conocimiento del comportamiento de un exón en las muestras controles como el caso del exón 19 en la familia del paciente 8, permite alcanzar a conclusiones con un grado muy alto de contabilidad.
En lo concerniente al consejo genético, los resultados permiten descartar, en gran medida, la posibilidad de que las mujeres a riesgo de ser portadoras de la deleción tengan una probabilidad significativamente aumentada de tener un hijo afectado por la misma mutación. No obstante, hay que tener en cuenta que las dosis génicas se evaluaron en tejido somático leucocitos; y que hay informes de que algunas mujeres pueden ser mosaicos (poseer células con la mutación y células normales) en sus tejidos germinales (23, 24).
Un hallazgo inesperado fue el hecho de que ninguna de las mujeres a riesgo portara las deleciones que afectan a los pacientes índice de sus respectivas familias. Esto se puede interpretar como que las mutaciones que los afectan surgen por primera vez, de novo, en sus familias, durante la oogenesis que produjo el óvulo del que ellos se originan. Según la literatura (25, 26) esto debería ocurrir en sólo un tercio de los casos. Aunque el número de familias estudiadas es pequeño -seis-, si esa proporción se cumpliera también en Costa Rica, la probabilidad de que todos los pacientes estudiados sean mutaciones de novo sería muy baja, sea de (1/3)6 = 1.37 x 10-3 . Este hallazgo señala un área de investigación muy interesante en epidemiología genética. Si lo observado en este estudio se confirmara habría una indicación de que la tasa de mutación en el gen de la distrofina es más alta que en otros países, como parece ser en el norte de la India (27). Esta posibilidad se podría probar mediante la identificación de un mayor número de pacientes con distrofinopatías, lo que además permitiría ampliar el tamizaje de deleciones y así aclarar otra aparente contradicción observada anteriormente (19), que sólo un 45% de los pacientes estudiados en Costa Rica presentan deleciones con respecto a lo descrito en otras partes del mundo (66%).
Esto se puede lograr únicamente con la colaboración de la comunidad médica nacional, en especial, la de los pediatras y neurólogos, si refirieran a este tipo de paciente a la Unidad de Genética y Metabolismo del Hospital Nacional de Niños. ]]>
Conclusiones
Todas las posibles portadoras mostraron dosis génicas que parecen compatibles con las de las mujeres sin deleciones. Esto añade más y mejores elementos de juicio para tomar en cuenta cuando se les brinde consejo genético. El resultado indica que todos los pacientes son producto de mutaciones de novo, lo que es inesperado, pues datos de otros países muestran que sólo 1/3 de los pacientes no heredan la mutación de madres portadoras
Agradecimientos
Al señor Víctor Castillo por su valioso trabajo técnico. A los Dres. Manuel Saborío, Carlos de Céspedes y Ramiro Barrantes por su apoyo en diversas etapas del trabajo. La investigación fue financiada por la Vicerrectoría de Investigación de la Universidad de Costa Rica, Proyecto742-97-253. Se contó también con la colaboración de FUCODOCSA
Referencias
1. Duchenne GBA. De I'electrisation localisée et son Application á la Pathologie et á la Thérapeutique. Paris: Bailliére et Fils, 1861. [ Links ]
2. Duchenne GBA. Recherches sur la paralyse musculaire pseudohypertrophique ou paralyse mio-sclerosique. Arch Gén Med 1868; 11:5-25, 179-209, 305-21, 421-43, 552-88. [ Links ]
3. Becker PE, Kiener F. Eine neue X- chomosomale Muskeldystrophie. Arch Psychiatr Nervenkrankheiten 1955; 193:427-48 [ Links ]
4. Emery AEH. Population frequencies of inherited neuromuscular diseases. A world survey. Neuromusc Disord 1991; 1:19-29. [ Links ]
5. OMIM (TM). Online Mendelian lnheritance in Man. Johns Hopkins University, Baltimore. http://www.ncbi.nlm.nih.gov/omim/, 2001 [ Links ]
6. Becker PIE. Dystrophia musculorum progressiva. Eine genetische und klinische Untersuchung der Muskeldystrophien.
Stuttgart: Georg Thieme, 1953
7. Emery AEH. Duchenne Muscular Dystrophy. Oxford University Press, Great Britain. 1993; 391 p. [ Links ]
8. Moser H, Emery AEH. The manifesting carrier in Duchenne muscular dystrophy. Clin Genet 1974; 5: 271-84. [ Links ]
9. Koenig M, Hoffman EP. Bertelson CJ, Monaco AP, Feener C, Kunkei LM. Complete cioning of the Duchenne muscular dystrophy (DMD) CDNA and preliminary genomic organization of the DMD gene in normal and affected individuas. Cell 1987; 50: 509-17. [ Links ]
10. Murray JM, Davies KE, Harper PS, Meredith L, Mueller CR, Williamson R. Linkage relationship of a cloned DNA sequence on the short arm of the X chromosome to Duchenne muscular dystrophy. Nature 1982; 300:69-71. [ Links ]
11. Ahn AH, Kunkei LM. The structural and functional diversity of dystrophin. Nature Genet 1993-1 3: 283-91. [ Links ]
12. Gillard EF, Chamberlain JS, Murphy EG, et al. Molecular and phenotypic analysis of patients with deletions within the deletion-rich region of the Duchenne muscular dystrophy (DMD) gene. Am J Hum Genet 1989-, 45: 507- 20. [ Links ]
13. Hu X, Ray P, Murphy G, Thompson M, Worton R. Duplicational mutation at the Duchenne muscular dystrophy locus: its frequency, distribution, origin, and phenotypegenotype correlation. Am J Hum Genet 1990; 46: 682-95. [ Links ]
14. Hiraishi Y, Kato S, lshihara T, Takano T. Quantitative Southern blot analysis of the dystrophin gene of Japanese patients with Duchenne or Becker muscular dystrophy: a high frequency of duplications. J Med Genet 1992; 29:897-901. [ Links ]
15. Tuffery S, Bareil C, Demaille J, Claustres M. Four novel dystrophin point mutations: Detection by protein truncation test and transcript analysis in lymphocytes from Duchenne Muscular Dystrophy patients. Eur J Hum Genet 1996; 4: 143-52. [ Links ]
16. Baxter PS, Maltby EL, Quarrell O. Xp2l Muscular dystrophy due to X chromosome inversion. Neurology 1997; 49: 260. [ Links ]
17. Chamberlain JS, Gibbs RA, Ranier JE. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucl Acids Res 1988; 16: 11141. [ Links ]
18. Beggs A, Koenig M, Boyce F, Kunkel L. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990; 86: 45-48. [ Links ]
19. Sancho-Fernández VM, Saborio M, de Céspedes C, Azofeifa J. Tamizaje de deleciones en pacientes con distrofina muscular de Duchenne (DMD) o de Becker-Kiener (BMD) mediante PCR múltiples en Costa Rica, 1998-2000. Acta Ped Cost. 2001 (enviado a publicación) [ Links ]
20. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 1988; 16:1215. [ Links ]
21. Yau SC, Bobrow M, Mathew CG, Abss SJ. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet 1996; 33: 550-9. [ Links ]
22. de Céspedes C, Umaña L, Yock I, Bustamante M, Atkins AT. Frecuencia y demanda de atención médica de las enfermedades genéticas en el Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera". Acta Ped Cost 1996; 10:53-60. [ Links ]
23. Bakker E, van Broeckhoven C, Bonten EJ, et al. Germline mosaicism and Duchenne muscular dystrophy mutations. Nature 1987; 329: 554-6. [ Links ]
24. Covone AE, Lerone M, Romeo G. Genotypephenotype correlation and germline mosaicism in DMD/BMD patients with deletions of the dystrophin gene. Hum Genet 1991; 87:353-60. [ Links ]
25. Emery AEH. Muscle histology and creatinine kinase levels in the foetus in Duchenne muscular dystrophy. Nature 1977; 266: 472-3. [ Links ]
26. Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet 1984; 66: 17-40. [ Links ]
27. Sinha S, Mishra S, Singh V, Mittal RD, Mittal B. High frequency of new mutations in North lndian Duchenne/becker muscular dystrophy patients. Clin Genet 1996; 50:327-31 [ Links ]
1Biólogos, genetistas, Instituto de Investigaciones en Salud (INISA) y Escuela de Biología, Universidad de Costa Rica
2Correspondencia: Dr. Jorge Azofeifa, Instituto de Investigaciones en Salud (INISA), Universidad de Costa Rica, Ciudad
Universitaria, San José, Costa Rica. Fax: ++506 2075130; correo electrónico: azofeifa@biologia.ucr.ac.cr ]]>
Abstract
Objective
To quantitate gene doses at the dystrophin locus in women at risk of being carriers of deletions.
Place where the work was performed: INISA and Escuela de Biología, Universidad de Costa Rica. Materiais and methods. Fifteen women were identified in a previous study to be at increased risk of being carriers of deletions at the dystrophin gene -they are first-degree relativas of patients suffering from DMD/BMD which owe their affection to deletions at the gene. DNAs of these women were used to amplify 7 to 9 regions of the gene by PCR-multiplex. To estímate gene doses the Fprimer of each pair was labeled with 6-FAM and the amplification products were separated and quantitated by fluorescent capillar electrophoresis.
Results
None of the 15 putative carriers showed evidence of carrying the deletions that affect the index patients in their families.
Conclusions
All index patients in the families of the carriers analyzed are affected by de novo mutations, an unexpected result according to data of other countries. The data add another, very valuable criterion to offer genetic counseling to the putative carriers.
Figura 1: Electroferogramas de los productos de amplificación de PCR-múltiplex usando 7 pares de imprimadores de una mujer control y tres pacientes afectados con diversas deleciones. Las flechas indican la ausencia de señales de aquellos exones que no amplificaron y por lo tanto se encuentran deletados. El panel lA corresponde a una mujer normal donde aparecen los picos (señales) de los siete exones que amplifican para este grupo de imprimadores, El panel lB pertenece a un paciente que presenta deleción de¡ exón 19, por lo que, está ausente el pico correspondiente a este exón. El panel 1C es de un paciente con una deleción que abarca los exones 6-42, por lo que no aparecen los picos de los exones 8, 12, 17 y 19. El panel 1D pertenece a un paciente afectado con la deleción de los exones 3-25, según se muestra, están ausentes los exones 4, 12, 8, 17 y 19. El área bajo los picos correspondientes a cada región amplificada es lo que sirve para cuantificar la dosis génica de las posibles portadoras. ]]>
]]>
