Frecuencia del alelo causante de la enfermedad de Charcot-Marie-Tooth (tipo axonal con herencia autosómica recesiva) en Palmares, Costa Rica
Melissa Rojas-Araya1, Roger Bonilla2, Sergio Campos3, Carolina Centeno4, Christian Del Valle1, Juan Pablo Pacheco3, Alina Saborío3, Jovel Vega3, Hubert Fernández5 & Alejandro Leal1
1. Escuela de Biología, Instituto de Investigaciones en Salud y Programa de Investigación en Neurociencias, Universidad de Costa Rica, 11501-2060, San José, Costa Rica. Fax: (506) 2511-4216; alejandro.leal@ucr.ac.cr
2. Centro Centroamericano de Población, Universidad de Costa Rica, 11501-2060, San José, Costa Rica.
3. Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica.
4. Departamento de Bioquímica, Escuela de Medicina, Universidad de Costa Rica, 11501-2060, San José, Costa Rica.
5. Servicio de Neurología, Hospital Rafael Ángel Calderón Guardia, Caja Costarricense de Seguro Social, San José, Costa Rica.
Abstract: Frecuency of the allele causing the axonal form of autosomal recessive Charcot-Marie-Tooth in Palmares, Costa Rica. The Charcot-Marie-Tooth disease constitutes is among the most frequent hereditary peripheral neuropathies world-wide. We identified a family from Palmares (Alajuela, Costa Rica) with 18 affected members. Their neuropathy is axonal, with an autosomal recessive pattern of inheritance; the responsible gene is at the 19q13.33 chromosomal region. Later the mutation was identified in gene MED25. We studied the frequency and geographic distribution of the mutant allele. In a random sample of 103 individuals, six were heterozygote and were widely distributed in Palmares. There was no person in homozigote state for the mutant allele. Clinical characteristics do not differ significantly between individuals that are homozygous for the wildtype allele and individuals hetero zygous for the mutation. A 5.83 % of the population is heterozygote and the frequency of the Ala335Val allele is 0.029, six times higher than in a sample of the Costa Rican population. Werecommend a molecular analysis of carriers to detect additional cases in the region. Rev. Biol. Trop. 57 (Suppl.1): 381-387. Epub 2009 November 30.
]]> Key words: Charcot-Marie-Tooth disease, CMT, HMSN, CMT2B2, Allelic frequency, Costa Rica.La enfermedad de Charcot-Marie-Tooth (CMT) también conocida como neuropatía hereditaria motora y sensorial (HMSN) abarca un grupo de trastornos que afectan los nervios periféricos (Suter & Scherer 2003) y es uno de los trastornos neurológicos hereditarios más comunes, pues afecta, por ejemplo, aproximadamente a 1 de cada 2 500 personas en los Estados Unidos (Reilly & Hanna 2002), tiene una prevalencia de 12.9-15.3 casos por cada 100 000 individuos en Cantabria, España (Combarros et al. 1987) y 20.1 casos por cada 100 000 personas en el norte de Suecia (Holmberg 1993). En Costa Rica no se conoce la prevalencia de esta neuropatía periférica.
CMT es causada por mutaciones dominantes o recesivas en genes que pueden ser expresados tanto en neuronas como en las células Schwann (Suter & Scherer 2003), los cuales codifican para proteínas relacionadas con la estructura y la función ya sea del axón del nervio periférico o de la capa de mielina.
Como características fenotipícas de la enfermedad se tienen las siguientes: debilitamiento en los pies, que pueden dar lugar a su deformación llamada pes cavus. Las deformidades del pie, tales como arcos altos y "dedos de martillo" (una condición en la cual la coyuntura central de un dedo del pie se dobla hacia arriba) son también características comunes (Pareyson 2004). Los pacientes también sufren de debilitamiento de los músculos distales de la pierna que genera una marcha a pasos grandes, además la parte inferior de las piernas puede adquirir un aspecto "de botella de champán invertida" debido a la pérdida de masa muscular (Tanaka & Hirokawa 2002). Conforme progresa la enfermedad, pueden ocurrir debilidades y atrofias musculares en las manos (Pareyson 2004).
De acuerdo a patrones clínicos, electrofisiológicos e histopatológicos, CMT se ha subdividido clásicamente en forma desmielinizante (CMT1) y forma axonal (CMT2). CMT1 es el tipo más frecuente y resulta principalmente de anormalidades en la capa de mielina. Se caracteriza por una velocidad de conducción nerviosa (por sus siglas en inglés NCV) de menos de 38 m/s, además estudios histológicos han revelado procesos de des y remielinación, así como también de la formación de bulbos tipo cebolla. CMT2 es menos frecuente, y se caracteriza por presentar valores de NCV mayores de 38 m/s, y valores muy disminuidos del potencial de acción evocado motor, (CMAP) y de amplitud del potencial de acción sensorial (SNAP) (Nicholson 2006). Estudio histopatológicos en biopsias han revelado atrofia y regeneración crónica en el axón (Gemignani & Martín 2001)
Hasta 1999 se conocían solamente tres loci relacionados con CMT autosómica dominante de tipo axonal (CMT2), y aún no se había identificado ningún locus relacionado con CMT autosómica recesiva de tipo axonal (CMT2B). Familias con este tipo de neuropatía suelen presentar un fenotipo clínico más severo y en algunos subtipos aparece muy temprano en la vida del paciente (Vallat et al. 2004). Sin embargo, hoy día ya se conocen tres loci responsables de CMT2B (Bouhouche et al. 2007). Bouhouche et al. (1999) estudiaron una familia consanguínea de origen Marroquí, con nueve individuos afectados que presentaban los síntomas clínicos clásicos para CMT, entre ellos una falla severa de los músculos proximales y datos electrofisiológicos indicaron un desorden axonal. Además, en dicha familia se seguía un patrón de herencia autosómica recesiva. Dichos autores encontraron evidencia de ligamiento con la región 1q21.2-q21.3 (CMT2B1). De Sandre-Giovanolli et al. (2002) reportan la mutación 892CT en el exón 5 del gen LMNA, el cual codifica para lamininas de la envoltura nuclear. Dicha mutación causa la substitución R238C y segregó con la enfermedad en trece individuos en familias de Argelia en los cuales se había encontrado ligamiento para la región 1q21.2-q21.3.
El segundo locus donde se encontró evidencia de ligamiento para CMT2B fue en la región cromosomica 8q21.3. Dicho estudio se realizo en trece pacientes procedentes de Túnez los cuales habían sido diagnosticados con debilitamiento distal progresivo de las extremidades y pérdida sensorial de las inferiores (Barhoumi et al. 2001). En 2002 Cuesta y colaboradores identificaron tres mutaciones y seis alelos mutantes en GDAP1 en tres familias con CMT axonal, con los cuales se había encontrado evidencia de ligamiento con la region 8q21.3. Dichos pacientes también presentaron paresis del cordón vocal. El gen GDAP1 se encuentra localizado en la región candidata, y se vio que su homólogo en ratón se encuentra altamente expresado en cerebro (Cuesta et al. 2002). GDAP1 es una proteína de 358 aminoácidos cuya función es desconocida, sin embargo estudios filogenéticos y análisis estructurales han sugerido que GDAP1 pertenece a la subfamilia de las glutation S-transferasas (GSTs) (Pedrosa et al. 2005)
El tercer locus fue reportado por Leal et al. (2001), ellos obvie en ligamiento significativo para CMT2B2 con la región cromosómica 19q13.3 en una familia consanguínea de la provincia de Alajuela, Costa Rica. Todos los pacientes fueron homocigotos y compartían el mismo haplotipo en la región. Los pacientes presentaron atrofias distales y debilitamiento muscular pronunciado y estudios electrofisiológicos demostraron un proceso axonal degenerativo, con CMAPs reducidos o ausentes. En dicho intervalo se identificaron 53 genes (Leal 2004), los cuales se secuenciaron en su totalidad. Posteriormente Leal y colaboradores localizaron en el exón 9 del gen MED25 la mutación Ala335Val y luego de probar 556 individuos control de la población costarricense, se propuso como causante de la patología (datos no pub.). Solamente los 18 individuos afectados de esta familia de Palmares han sido encontrados como homocigotos para el alelo Ala335Val, y de los 556 controles, cinco fueron heterocigotos, por lo que la frecuencia del alelo mutante en la población general es de q=0.0045. El gen MED25 codifica para la subunidad 25 del complejo mediador de la transcripción por ARN polimerasa II, dicho complejo mediador actúa como un coactivador clave en la activación de la trascripción (Malik 2005). La mutación Ala335Val realiza un cambio de aminoácido en una zona proteica muy conservada. Recientemente se han hecho numerosos estudios de función de la proteína solidificándose la evidencia de que se trata de la responsable de la enfermedad (Leal et al., datos no pub.). No obstante, todavía se desconoce cuál es el rol de MED25 en la patología CMT2B2.
Como se indicó anteriormente, se desconoce cuál es la prevalencia de la enfermedad de Charcot-Marie-Tooth en la población de Costa Rica. Además se desconoce cuál es la prevalencia de la enfermedad en el cantón de Palmares de Alajuela de donde procede la familia afectada, así como la frecuencia del alelo mutado en dicha población. Existe la necesidad de determinar la frecuencia de la mutación Ala335Val en el gen MED25 en la población de donde se origina la familia afectada con CMT2B2, así como establecer la prevalencia de CMT en la población de donde se origina la familia afectada.
Materiales y métodos
]]> La población de estudio fue la población del cantón de Palmares Alajuela, Costa Rica; lugar donde se localizó la familia que permitió identificar el locus CMT2B2. Se tomó una muestra representativa y aleatoria de 103 personas de dicha población alajuelense, tomando en cuenta los siete distritos del cantón. Se calculó el tamaño de muestra asumiendo muestreo simple aleatorio (MIA), ya que se desconoce la distribución de la incidencia de la enfermedad en el cantón de Palmares. Según el Censo de Población 2000, la población del cantón es de cerca de 30 mil personas. Se asumió que la dispersión de la enfermedad era máxima (no concentración de la enfermedad en conglomerados geográficos en la zona). Lo anterior significa que la variancia de la proporción de personas con la enfermedad era máxima y eso se da cuando la proporción ~ p es igual a 0.5, entonces la variancia estimada es igual a ~ p (1 - ~ p) = 0.25. El error máximo permisible d se fija es 0.1, o sea 10 %, esto significa que la estimación obtenida en la encuesta de personas infectadas con la enfermedad no superará en 0.1 del valor que se obtendría si re realizara un censo general en Palmares, lo cual es impensable. Se estimó que la muestra apropiada es de 76 personas. Sin embargo, se entrevistaron 103 personas pues existían los recursos disponibles. El ajuste por población finita no da valor agregado significativo. La muestra fue distribuida proporcionalmente entre los distritos por tamaño de población.A cada una de las personas de la muestra se le tomó información personal, familiar, genealógica y se le hizo una evaluación clínica, luego de obtener el consentimiento informado correspondiente. La examinación clínica consistió en pruebas de fuerza y sensibilidad muscular estandarizadas (Berghoff et al. 2004). Por dificultades técnicas no se hicieron mediciones electrofisiológicas. A cada individuo se le tomo 5 mL de sangre periférica, la extracción de ADN se hizo mediante el kit de Purificación de ADN Genómico (Fermentas). Se amplificó por PCR la región donde se encuentra la mutación Ala335Val en MED25, utilizando iniciadores (primers) específicos, primer F:5´- CAGAACCACGAGAGCCACATCC-3´ primer R:5´-GTCCCCCAGAGCCCCAT CAAC-3´, Taq polimersa y reactivos para PCR marca Fermentas siguiendo las recomendaciones del fabricante.
Para confirmar la ampliación por PCR se corrieron los productos por geles de acrilamida 10%. Una vez amplificado el segmento específico, los productos de PCR se digirieron con enzimas de restricción, incubando las muestras a 37 °C toda la noche. Las enzimas de restricción utilizadas son NarI y HaeII que cortan la secuencia silvestre, y HgaI que corta la secuencia con la mutación. Posteriormente se realizó una segunda electroforesis con geles de acrilamida 10% para poder visualizar los fragmentos digeridos y conocer así el estatus genético de cada individuo de la muestra. Además, se cargó un control positivo y negativo de corte tanto para NarI como para HaeII, se contó con pacientes de estudios anteriores los cuales se les había confirmado la presencia o ausencia de la mutación por secuenciación.
Se utilizó el programa GenAlEx 6.0 para calcular las frecuencias alélicas, las frecuencias genotípicas y la heterocigosis. Los cálculos fueron realizados con el programa STATA versión 8 (StataCorp 2005) y el nivel de significancia de las pruebas se fijó en un α =0.05. Se utilizó la prueba Chi cuadrado para medir asociaciones entre variables categóricas (Agresti 2002) y la prueba de t-Student para comparar promedios.
Resultados
Además de la familia afectada con CMT2B2 (Leal et al. 2001), ningún individuo enfermo adicional se localizó en este rastreo. En la muestra sí apareció asignado un individuo con familiares afectados, pero todos pertenecían a la familia con 18 casos que había servido para hacer la búsqueda genómica y el análisis de ligamiento.
En cuanto a las pruebas moleculares, los resultados de la digestión con enzimas de restricción indicaron que de las 103 muestras 97 personas son homocigotas silvestres y seis heterocigotas para la mutación. En la distribución geográfica del alelo Ala335Val no se observa ninguna agregación de los heterocigotos por distrito. No se encontró ninguna persona en estado homocigota para el alelo Ala335Val en la muestra analizada. El porcentaje de la población que es homocigota silvestre y heterocigota para Ala335Val es del 94.17% y 5.83% respectivamente. Además, la frecuencia del alelo silvestre en el cantón es de 0.971 y la frecuencia del alelo Ala335Val es de 0.029. Las frecuencias genotípicas se encontraron en equilibrio de HW (Chi=0.094, gl=1, p=0.760). Asimismo se obtuvo un valor de He de 0.057.
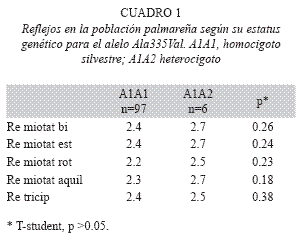
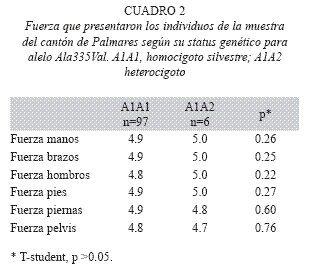
El análisis estadístico permitió determinar que entre los individuos homocigotos silvestres y los heterocigotos para la mutación no existe ninguna característica clínica que difiera significativamente entre ambos. No obstante, se observaron pequeñas diferencias en los reflejos y en la fuerza. Se halló que los reflejos en los individuos heterocigotos se encontraban aumentados con respecto a los individuos homocigotos silvestres (Cuadro 1). También se observo que la fuerza en la pelvis y en las piernas estaba disminuida en los individuos heterocigotos, ninguno de los dos resultados fue significativo (Cuadro 2).
De los seis individuos heterocigotos para Ala335Val, solo un individuo tiene antecedentes familiares de CMT, con tres parientes (tíos y padre) que padecen de la enfermedad. A uno de dichos pacientes se le hizo el análisis clínico y genético para tener un control positivo en el estudio, sin embargo dicho paciente no fue tomado en cuenta en el análisis estadístico ya que no fue parte de la muestra. El paciente presentó corte positivo con HgaI y corte negativo para NarI, por lo que el individuo es homocigoto para Ala335Val. Los datos clínicos mostraron que el paciente tiene reflejos y sensibilidad muy disminuidos, casi nulos. Además, presentó dedos de martillo, tremor, atrofias tanto en manos como en piernas. Los problemas sensoriales que presentó el paciente fueron principalmente en manos, ya que presentó adormecimiento y sensaciones anormales espontáneas.
Discusión
Una familia de un cantón de Palmares de la provincia de Alajuela sirvió para determinar ligamiento entre una forma axonal de la enfermedad de Charcot-Marie-Tooth con la región cromosómica 19q13.33, y posteriormente se localizó en esa región la mutación Ala335Val causante de la patología en MED25 (Datos no pub.). Este hallazgo sirvió para realizar un estudio epidemiológico para determinar la prevalencia de la enfermedad en el cantón, así como la frecuencia de la mutación en la población.
En el presente estudio se determinó que la frecuencia del alelo Ala335Val en el cantón en estudio es de 0.029, 6.45 veces mayor que en la población general de Costa Rica, observándose una heterocigosis de 0.057. Dado, quela heterocigosis es una medida de la variación genética de un locus en una población, si la frecuencia de uno de los alelos es muy alta y la otra muy baja, habrán pocos heterocigotos porque la mayoría de los individuos serán homocigotos para el alelo más frecuente. Esto ocurre en este estudio, en el cual el alelo más frecuente es el alelo homocigoto silvestre.
]]> No se encontraron pacientes nuevos de CMT2B2 en la población muestreada, lo que nos podría indicar que la enfermedad esta segregando exclusivamente en las familias donde se halló ligamiento. Solamente los 18 individuos afectados reportados por Leal et al. (2001) han sido encontrados como homocigotos para el alelo mutado.Según el último censo poblacional que se hizo en nuestro país en el 2000, se obtuvo el cantón de Palmares tiene alrededor de 30000 habitantes. En este estudio se encontró que el 5.83 % de la población es heterocigoto por lo que se podría estimar que en el cantón en estudio hay alrededor de 1 700 individuos heterocigotos para Ala335Val.
Lo anterior se acentúa cuando hay cruces consanguíneos en la población ya que dichos cruces aumentan la homocigosis y disminuye la heterocigosis en las frecuencias genotípicas. En Marruecos se ha visto que CMT con herencia autosómica recesiva es más frecuente que la forma dominante de la enfermedad, esto se debe a que en el norte de África los matrimonios consanguíneos son comunes (Bouhouche et al. 2007).
Clínicamente los individuos heterocigotos no mostraron ninguna diferencia significativa a los individuos homocigotos silvestre, lo cual era de esperar al ser CMT2B2 una enfermedad autosómica recesiva. Los que los individuos heterocigotos no padecen de la enfermedad pero si son portadores del alelo Ala335Val, y habría que hacer estudios con una muestra mayor para observar si existen características clínicas significativamente distintas entre heterocigotos y homocigotos silvestres.
Este estudio debe ser considerado como un estudio piloto, ya que solo se muestreó el cantón donde fue descrita la mutación. Sería interesante conocer la frecuencia del alelo Ala335Val en cantones cercanos al cantón estudiado y hasta se podría pensar en hacer un muestreo aleatorio de toda la provincia de Alajuela y en el país. De este modo, se podrían obtener más datos que nos permitan la detección de nuevos casos de afectados, que aumenten la información genética y clínica de la enfermedad CMT2B2, y se podría proporcionar a los individuos afectados más información sobre su padecimiento.
La frecuencia de heterocigotos (portadores) del alelo mutado sugiere que un diagnóstico molecular de los portadores se debería ofrecer a la población del cantón de Palmares, con el fin de que este conocimiento sea tomado en cuenta por las personas interesadas antes de asumir la responsabilidad de reproducirse. Este es el primer estudio epidemiológico de la enfermedad de Charcot-Marie-Tooth en Costa Rica, y uno de los primeros en enfermedades hereditarias. Estudios sucesivos deberían permitir mayor conocimiento de las enfermedades con un componente genético importante, favoreciendo su prevención y tratamiento.
Agradecimientos
Este estudio fue financiado por la Vicerrectoría de Investigación de la Universidad de Costa Rica (Proyecto No. 742-98-241). Agradecemos a la población del cantón de Palmares por la buena voluntad al participar en el estudio, muy especialmente a los 103 individuos muestreados. Agradecemos, además, a Carolina Araya Callís y a Omar Achí Corrales por la ayuda técnica.
Resumen
]]> La enfermedad de Charcot-Marie-Tooth constituye elgrupo de neuropatías periféricas hereditarias más común a nivel mundial. Una familia con 18 afectados del cantón de Palmares (Alajuela, Costa Rica) con una neuropatía de tipo axonal y herencia autosómica recesiva, permitió localizar el gen responsable en la región 19q13.33. Posteriormente se identificó la mutación causante en el gen MED25. El presente estudio determinó la frecuencia del alelo mutante, así como la distribución geográfica de este alelo. En una muestra al azar de 103 individuos se encontraron seis individuos heteroigotas para la mutación, distribuidos por todo el cantón. No se encontró ninguna persona en estado homocigota para este alelo. No hallamos algunacaracterística clínica que difiera significativamente entre los individuos homocigotos silvestres y los heterocigotos para la mutación. El 5.83% de la población es heterocigota y la frecuencia del alelo Ala335Val es de 0.029, seis veces mayor que en una muestra de toda la población costarricense. Por esta razón se recomienda un análisis molecular de portadores con el fin de alertar sobre la posibilidad de aparición de más casos en el cantón.Palabras clave: enfermedad de Charcot-Marie-Tooth, CMT, HMSN, CMT2B2, frecuencia alélica, Costa Rica.
Recibido 03-X-2007. Corregido 01-IX-2009. Aceptado 16-X-2009.
Referencias
Agresti, A. 2001. Categorical Data Analysis. John Wiley & Sons, Nueva Jersey, EEUU. [ Links ]
Barhoumi, C., R. Amouri, C. Ben Hamida, M. Ben Hamida, S. Machghoul, M. Gueddiche & F. Hentati. 2001. Linkage of a new locus for autosomal recessive axonal form of Charcot-Marie-Tooth disease to chromosome 8q21.3. Neuromuscular Disord. 11: 27-34. [ Links ]
Berghoff, C., M. Berghoff, A. Leal, B. Morera, R. Barrantes, A. Reis, B. Neundoerfer, B. Rautenstrauss, G. Del Valle & D. Heuss. 2004. Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (CMT2B2) that maps to chromosome 19q13.3. Neuromuscular Disord.14: 301-306. [ Links ]
Bouhouche, A., A. Benomar, N. Birouk, A. Mularoni, F. Meggouh, J. Tassin, D. Grid, A. Vandenberghe, M. Yahyaoui, T. Chkili, A. Brice & E. LeGuern. 1999. ALocus for an Axonal Form of Autosomal Recessive Charcot-Marie-Tooth. Disease Maps to Chromosome 1q21.2-q21.3. Am. J. Hum. Genet. 65: 722-727. [ Links ]
Bouhouche, A., N. Birouk, H. Azzedine, A. Benomar, G. Durosier, D. Ente, M.P. Muriel, M. Ruberg, I. Slassi, M. Yahyaoui, O. Dubourg, R. Ouazzani & E. LeGuern 2007. Autosomal recessive axonal Charcot Marie Tooth disease (CMT2B): phenotype genotype correlations in 13 Moroccan families. Brain 130: 1062-1075. [ Links ]
Combarros, O., J. Calleja, J.M. Polo & J. Berciano. 1987. Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol. Scand. 75: 9-12. [ Links ]
Cuesta, A., A. Pedrola, T. Sevilla, J. García-Planells, M.J. Chumillas, F. Mayordomo, E. LeGuern, I. Marín, J.J. Vílchez & F. Palau. 2002. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet. 30: 22-25. [ Links ]
De Sandre-Giovannoli, A., M. Chaouch, S. Kozlov, J.M. Vallat, M. Tazir, N. Kassouri, P. Szepetowski, T. Hammadouche, A. Vandenberghe, C.L. Stewart, D. Gris & N. Levy. 2002. Homozygous defects in LMNA, encoding lamin a/c nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (charcot-marie-tooth disorder type 2) and mouse. Am. J. Hum. Genet. 70: 726-736. [ Links ]
Holmberg, B. 1993. Charcot-Marie-Tooth disease in northern Sweden : an epidemiological and clinical study. Acta Neurol. Scand. 87: 416-422. [ Links ]
Instituto Nacional de Estadística y Censos. 2001. IX Censo Nacional de Población y V de Vivienda del 2000: Resultados Generales. Instituto Nacional de Estadística y Censos. Instituto Nacional de Estadísticas y Censos, San José, Costa Rica. [ Links ]
Gemignani, F. & A. Marbini. 2001. Charcot-Marie-Tooth disease (CMT): distinctive phenotypic and genotypic features in CMT type 2. J. Neurol. Sci. 184: 1-9. [ Links ]
Pareyson, D. 2004. Differential diagnosis of Charcot- Marie-Tooth disease and related neuropathies. Neurol.Sci. 25: 72-82. [ Links ]
Leal, A. 2004. Genetics of hereditary motor and sensory neuropathy and the Costa Rican contribution. Rev. Biol. Trop. 52: 475-483. [ Links ]
Leal, A., B. Morera, G. Del Valle , D. Heuss, C. Kayser, M. Berghoff, R. Villegas, E. Hernandez, M. Mendez, H.C. Hennies, B. Neundoerfer, R. Barrantes, A. Reis & B. Rautenstrauss. 2001. A second locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 19q13.3. Am. J. Hum. Genet. 68: 269-274. [ Links ]
Malik, S. & R. Roeder. 2005. Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem. Sci. 30: 256-263. [ Links ]
Nicholson, G.A. 2006.The dominantly inherited motor and sensory neuropathies: clinical and molecular advances. Muscle Nerve. 33: 589-597. [ Links ]
Reilly M.M. & M.G. Hanna. 2002. Genetic neuromuscular disease. J. Neurol. Neurosurg. Psychiatry 73: 12-21. [ Links ]
StataCorp. 2005. Stata Statist. Software: Release 8. College Station, Texas, StataCorp LP, Texas, EEUU. [ Links ]
Suter, U. & S.S. Scherer. 2003. Disease mechanisms in inherited neuropathies. Nat. Rev. Neurosci. 4:714-726. [ Links ]
Tanaka Y & N. Hirokawa. 2002. Mouse models of Charcot- Marie-Tooth disease. Trends Genet. 18: S39-S44. [ Links ]
Vallat, J.M., D. Grid, C. Magdelaine, F. Sturtz & M. Tazi. 2004. Autosomal Recessive Forms of Charcot-Marie-Tooth Disease. Curr. Neurol. Neurosci. Rep. 4: 413-419. [ Links ]
]]>