Caso Clínico
Mucopolisacaridosis II: nueva mutación patogénica en gen IDS
Mucopolysaccaridosis II: new pathogenic mutation in IDS gene
Eugenia Pérez-Elizondo1* Alejandra Acosta-Gualandri2* y Mildred Jiménez-Hernández3*
La mucopolisacaridosis tipo II es una enfermedad lisosomal producida por la deficiencia de la enzima iduronato 2 sulfatasa. Es una condición infrecuente de herencia recesiva ligada al X, que puede producir importante discapacidad progresiva. El análisis molecular es una técnica útil en la confirmación diagnóstica, que además permite detección de portadores asintomáticos, brindando la oportunidad de asesoría genética. Se presenta el caso de un paciente con mucopolisacaridosis tipo II, en quien se documentó una nueva mutación patogénica en el Gen IDS.
Descriptores: mucopolisacaridosis, síndrome de Hunter, enfermedad lisosomal, iduronato-2-sulfatasa, glicosaminoglicanos
Abstract
Keywords: mucopolysaccaridosis, Hunter Syndrome, lysosomal disease, iduronate-2-sulphatase enzyme, glycosaminoglycans.
La mucopolisacaridosis tipo II (síndrome de Hunter) se produce por una deficiencia de la enzima Iduronato 2 sulfatasa, encargada de la degradación de los glicosaminoglicanos dermatán sulfato y heparán sulfato.1,2 Es de herencia recesiva ligada al X, ya que el gen IDS, que codifica por Ia enzima Iduronato 2 Sulfatasa, se encuentra en el cromosoma Xq28.1,2,3 Lo anterior explica porque la mayoría de casos son del sexo masculino.
Al nacimiento pueden tener una apariencia normal y presentar adecuado desarrollo psicomotor, sin embargo, con el depósito progresivo de glicosaminoglicanos, se presenta gran afección multisistémica y alto grado de discapacidad. Para su diagnóstico y manejo es necesario un enfoque multidisciplinario y oportuno.
Caso clínico
Al examen físico se observa un paciente con baja talla, cuello corto, quien presenta limitación para la flexión y extensión cervical, pabellones auriculares gruesos, nariz bulbosa, labios y encías engrosadas. Tórax corto y prominente, ruidos cardiacos rítmicos con soplo sistólico grado I/VI. Abdomen globoso, hígado palpable a
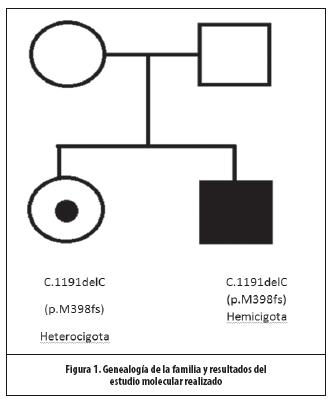
Ultrasonido de abdomen evidencia hepatoesplenomegalia con esteatosis hepática leve. Hipoacusianeurosensorial levey glaucoma bilateral. Patrón pulmonar restrictivo y asma persistente en control por Neumología. Ecocardiograma con insuficiencia mitral y aórtica leve, con engrosamiento valvular. Médula ósea normal. Se documentó una elevación anormal de mucopolisacáridos en orina, y niveles indetectables de la enzima Iduronato 2 sulfatasa, al realizar análisis enzimático. Se obtuvo por medio de análisis molecular del gen IDS, la presencia de la mutación c.1191delC (p.M398fs) en estado hemicigoto. (Figura 1)
Discusión
El reconocimiento del fenotipo del paciente con mucopolisacaridosis (MPS) tipo II permite el diagnóstico diferencial con los diferentes subtipos de MPS y otras enfermedades de depósito lisosomal, orientando al clínico a los estudios confirmatorios.
La apariencia facial es tosca por la infiltración de glicosaminoglicanos (GAG) en tejido conectivo de la región oro facial y es aparente a la edad promedio de 3 años.1,2 Hay engrosamiento del cuero cabelludo con hipertricosis y lesiones papulares perladas en escápulas, tronco y muslos.1
Se produce una poliartropatía destructiva que afecta principalmente las caderas. Las manos desarrollan una deformidad en garra con síndrome de túnel carpal y limitación motora por rigidez de las articulaciones interfalángicas.1,2
Es frecuente la valvulopatía mitral y aórtica por engrosamiento valvular, con mayor incidencia de cardiomiopatía hipertrófica asociada a arritmias.1,2,3,4 La infiltración del tejido respiratorio por GAG produce engrosamiento de adenoides, amígdalas, tráquea, cuerdas vocales y lengua, causando crisis de apneas obstructivas con hipoxemia crónica y mayor dificultad para intubación endotraqueal.1,2,5
Las hernias por debilidad en la pared abdominal son frecuentes, así como la hepatoesplenomegalia sin repercusión funcional.1,2
Se ha reportado degeneración retiniana, sin embargo, el glaucoma y la opacidad corneal son inusuales en MPS II.1
La hipoacusia es común y se correlaciona con lo severo de la enfermedad. Inicialmente es conductiva y luego desarrolla un componente neurosensorial.1,6
Entre los estudios bioquímicos preliminares está la medición de MPS en orina, la cual puede ser cuantitativa o cualitativa. Es una prueba útil, porque ante la sospecha de una enfermedad de depósito, la elevación de MPS tiene un valor predictivo positivo.7 Sin embargo, una prueba negativa no descarta el diagnóstico, ya que la excreción renal de MPS varía en función inversa a la edad del niño, siendo mayor durante los primeros años de vida y disminuyendo hasta estabilizarse a los 9 años.8 La medición cualitativa determina qué tipo de sulfato se encuentra elevado en orina, y esto ayuda a categorizar el diagnóstico de MPS II sin confirmarlo, ya que las MPS I y III pueden presentar el mismo patrón de sulfatos en la orina.7
En el caso, el Laboratorio de Bioquímica de Alto Riesgo del Hospital Nacional de Niños realizó una medición cuantitativa de MPS en orina, utilizando la técnica de Hawksworth modificada por Pennock. El paciente presentó 520 mg de MPS en orina/gr de creatinina, el cual es elevado.
El diagnóstico definitivo amerita ensayos enzimáticos en plasma, leucocitos o fibroblastos, utilizando sustratos específicos para Iduronato 2 Sulfatasa (I2S).1,2 La ausencia o disminuida cantidad de I2S en pacientes masculinos, es diagnóstica de MPS II, pero el grado de actividad no predice la severidad del fenotipo y no permite la detección de portadoras femeninas.7 Se envió una muestra de sangre periférica al Laboratorio de Neurometabolismo Dr. N.A. Chamoles, en Buenos Aires, Argentina, reportándose niveles indetectables de la enzima I2S. Lo anterior confirmó el diagnóstico.
El análisis molecular es útil para la confirmación diagnóstica, pero además es la única forma segura de detectar portadoras.7 Su utilidad ha sido descrita también en estudios prenatales, sin embargo, no es una técnica siempre accesible, y existen casos en los que no se puede detectar la mutación.1,2
La terapia de remplazo enzimático ha sido aprobada para su uso en los Estados Unidos y la Unión Europea. Los resultados aún no son concluyentes, pero se reporta mejoría en la movilidad articular, función pulmonar, tolerabilidad para actividad física, así como reducción del tamaño del hígado y bazo, sin observarse mejoría en el sistema nervioso central.1,2
Debido a que en nuestro medio aún no se cuenta con este tratamiento, no fue posible la implementación del remplazo enzimático.
El trasplante de médula ósea es otra opción terapéutica que se ha planteado y que ha reportado resultados favorables en ciertos pacientes.1,10
Aunque la MPS II es infrecuente y las opciones terapéuticas que modifiquen significativamente su curso son limitadas, es importante el diagnóstico temprano que permita brindar al paciente tratamiento sintomático que mejore su calidad de vida, así como asesoría genética al núcleo familiar, posibilitando tomar una decisión reproductiva informada.
Referencias
1. Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:3:267-77. [ Links ]
2. Guelbert N, Amartino H, Arberas C, Azar N, Bay L, Fainboim A, et al. GUideline for diagnosis, follow-up and treatment of mucopolysaccharidoses type II or Hunter disease. Arch Argent Pediatr. 2011;109:2:175-81. [ Links ]
3. Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol 2005,32:270-2. [ Links ]
4. Rigante D, Segni G. Cardiac structural involvement in mucopolysaccharidoses. Cardiology.2002;98:18-20. [ Links ]
5. Leighton SE, Papsin B, Vellodi A, Dinwiddie R, Lane R. Disordered breathing during sleep in patients with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2001;58:127-38. [ Links ]
6. Simmons MA, Bruce IA, Penney S, Wraith E, Rothera MP. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2005; 69:589-95. [ Links ]
7. Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Muñoz V et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter Syndrome). Pediatr. 2008;121:377. [ Links ]
8. Apéstegui A, Trejos R, Blanco A, González L. Valores normales de mucopolisacáridos en orina de niños. Rev. Med. Hosp. Nal. Niños Costa Rica. 1980;215-224. [ Links ]
9. Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2013. En: www.hgmd.cf.ac.uk. [ Links ]
10. Malatack JJ, Consolini DM, Bayever E. The status of hematopoietic stem cell transplantation in lysosomal storage disease. Pediatr Neurol. 2003; 29:391-403. [ Links ]
Trabajo realizado en: Hospital Nacional de Niños Dr. Carlos Sáenz Herrera
Afiliación de los autores: 1,2Servicio de Genética Médica y Metabolismo,3División de Genética Molecular, Laboratorio Nacional de Tamizaje Neonatal y Alto Riesgo, Hospital Nacional de Niños. eu-perezelizondo@hotmail.com
Fecha recibido: 06 de enero de 2014 Fecha aprobado: 19 de junio de 2014
{kind=link}