Caso clínico
Distrofia muscular de Duchenne y defecto de oxidación de ácidos grasos en un paciente pediátrico
Duchenne muscular dystrophy and fatty acid oxidation defect in a pediatric patient
Alejandra Acosta-Gualandri1* y Mildred Jiménez-Hernández2*
Resumen
El paciente presenta una enfermedad metabólica debido a un trastorno en la oxidación mitocondrial de los ácidos grasos, con una clínica caracterizada por hipoglicemias no cetósicas, cuya herencia es autosómica recesiva, así como una condición neurodegenerativa, que obedece a un defecto en la síntesis de la proteína de la distrofina, con una herencia ligada al X de forma recesiva. Ambas enfermedades genéticas son poco frecuentes, con patrones de herencia mendeliana diversos, presentes en un mismo individuo.
Descriptores: déficit de acil–CoA deshidrogenasa, ácidos grasos, defecto de betaoxidación, distrofia muscular Duchenne, distrofina
Abstract
Clinical presentation of a pediatric patient diagnosed with Duchenne muscular dystrophy and medium chain acyl-CoA dehydrogenase deficiency. Both diseases were confirmed by a molecular analysis that detected the deletion of exons 45 to
The patient has a metabolic disease due to a mitochondrial fatty-acid oxidation disorder, characterized by non-ketotic hypoglycemias, with an autosomal recessive inheritance and a neurodegenerative condition, due to a defect in dystrophin protein synthesis, with an X- linked recessive inheritance. These rare genetic diseases, with different Mendelian inheritance patterns are present in the same individual.
Keywords: acyl-CoA dehydrogenase deficiency, fatty acids, beta oxidation disorder, Duchenne muscular dystrophy, dystrophin.
La distrofia muscular de Duchenne (DMD) es una enfermedad genética neurodegenerativa, caracterizada por debilidad muscular progresiva. Afecta principalmente a hombres, con una incidencia de 1 de cada 3600-6000 varones nacidos vivos.1,2 Presenta una herencia ligada al cromosoma X de forma recesiva y obedece a la mutación del gen encargado de la síntesis de distrofina, proteína esencial del tejido neuromuscular. La deficiencia de acil-CoA deshidrogenasa de cadenas medias, es un defecto en la oxidación de ácidos grasos (DOAG) a nivel mitocondrial. Afecta tanto hombres como mujeres, con una incidencia de 1 de cada 4900 - 17000 individuos.3 Su herencia es autosómica recesiva, con una presentación clínica variable, desde sintomatología leve hasta la muerte súbita. Se caracteriza por la presencia de hipoglicemias no cetósicas en los periodos de ayuno.
Las enfermedades monogénicas, como las mencionadas, resultan de alteraciones en la secuencia de ADN de un solo gen; son condiciones relativamente poco frecuentes, que afectan a un porcentaje muy pequeño de la población mundial. La presencia de dos enfermedades monogénicas, que involucran genes diferentes, con patrones de herencia mendeliana diversa, convierte a nuestro paciente en un caso único.
Presentación de caso clínico
Paciente masculino que a los 15 días de edad fue referido por el Programa Nacional de Tamizaje, debido a elevación de los metabolitos C6 y C8 en el perfil de acilcarnitinas de la prueba de talón; fue valorado por el Servicio de Genética y Metabolismo del Hospital de Niños, donde se realizaron estudios por la sospecha de un defecto de oxidación de ácidos grasos (DOAG).
Hijo de padres consanguíneos. Sin antecedentes perinatales de importancia, con un examen físico normal. No se documentó acidosis metabólica, hipoglicemia o cetosis. En el análisis de ácidos orgánicos en orina, presentó importante elevación de hexanoilglicina, elevación moderada de ácido subérico y de suberilglicina, así como ligera elevación de ácido 7-OH-caprilico y ácido 5-OH-caproico, relacionados con la deficiencia de acil-CoA deshidrogenasa de cadenas medias. El análisis molecular del gen ACADM confirmó el diagnóstico al detectarse la mutación A985G en estado homocigoto.
A la edad de 4 años, el paciente presentaba fatiga, mialgias y pérdida de las habilidades motoras previamente adquiridas. Se documentó bajo peso y talla, debilidad muscular, limitación para deambular, signo de Gowers positivo, pseudohipertrofia de gastronemios y cifoescoliosis tóracolumbar.
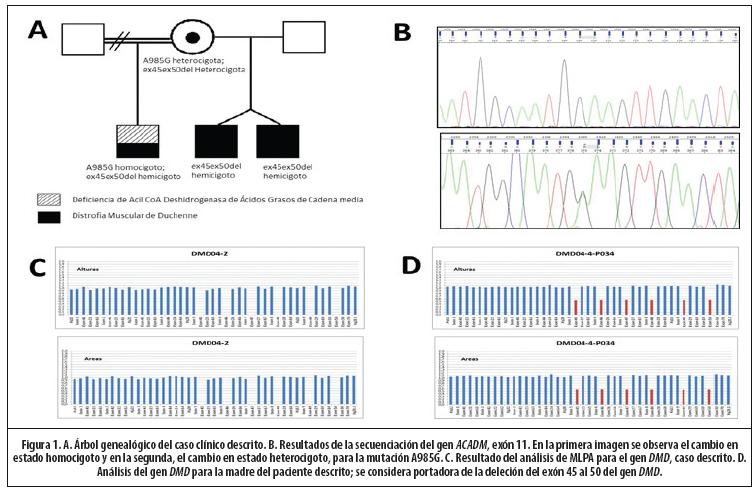
El paciente se encuentra en tratamiento con esteroides, mantiene un plan nutricional adecuado, asiste a fisioterapia y a la escuela de enseñanza especial. Cabe mencionar que tiene 2 medios hermanos varones, gemelos. Ambos, presentaron deleciones en los exones del 45 al 50, por lo cual también padecen de DMD. En ninguno se detectó la deficiencia de acil-CoA deshidrogenasa de cadenas medias. La madre de estos niños es portadora para la deleción de los exones 45-50 en el gen DMD, y para la mutación A985G en el gen ACADM. (Figura 1)
Discusión
La DMD es una enfermedad neuromuscular hereditaria, caracterizada por debilidad muscular progresiva.1 Presenta una herencia ligada al X de forma recesiva, por lo cual afecta predominantemente a hombres. Solo un 10% de las mujeres portadoras tiene alguna manifestación clínica, ya que las sintomáticas son el resultado de la inactivación del cromosoma X sano.4
A nivel mundial afecta a 1 de cada 3600-6000 varones nacidos vivos.1,2,4 Sin embargo, en Costa Rica no se cuenta con datos sobre la incidencia o frecuencia génica de esta enfermedad. Otros países, como Canadá, han reportado una incidencia de 1 en 4700 varones nacidos vivos; en Inglaterra, de 1 de cada 18450 varones se ve afectado por esta enfermedad, y en Noruega, se presenta en 1 de cada 3917 varones nacidos vivos.2
La distrofina se encuentra en el tejido muscular cardiaco, esquelético y liso, y en pocas cantidades en el sistema nervioso central y periférico.1 Esta proteína es parte importante de las membranas plasmáticas y de las uniones miotendinosas y neuromusculares.1
La distrofina es una proteína del citoesqueleto, la cual se une a la actina e interactúa con un complejo glicoproteína-distrofina (CGD), que une citoesqueleto, lámina basal y membrana plasmática.1,5,6 En la biopsia de músculo de un paciente con DMD, la distrofina está ausente del musculo esquelético, y las fibras musculares se observan necróticas o degeneradas. Las manifestaciones clínicas en dichos individuos, son consideradas el resultado de un desbalance entre la necrosis de las fibras musculares y la regeneración mioblástica.6,7
El diagnóstico se realiza al observar un deterioro progresivo y simétrico de la fuerza muscular proximal, limitación para deambular, marcha de puntillas, signo de Gower positivo e hipertrofia de pantorrillas.4 Los problemas de la columna vertebral, como la escoliosis y cifosis, pueden limitar la mecánica pulmonar y producir afección del musculo cardiaco.4 Generalmente, estos pacientes fallecen debido a complicaciones respiratorias o cardiacas. 1
La otra condición identificada en el paciente, es un defecto en la enzima acil-CoA deshidrogenasa de cadenas medias, una de las encargadas de la betaoxidación de ácidos grasos a nivel mitocondrial. 8
Los ácidos grasos son utilizados por el músculo cardiaco y esquelético, como principal fuente de energía durante el ejercicio, y durante el ayuno prolongado, por la mayoría de los tejidos.8
La deficiencia de acil- CoA deshidrogenasa de cadenas medias, se observa en individuos de ascendencia europea. Se estima que la frecuencia mundial se encuentra entre 1:4900 y 1:17000, la variabilidad en dichas cifras se relaciona con el origen étnico de la población estudiada. El número de recién nacidos con la deficiencia enzimática detectados mediante programas de tamizaje neonatal, es superior a la esperada con base en la frecuencia de la población. Para Costa Rica, se estima, según datos no publicados del Programa Nacional de Tamizaje, que la frecuencia de aparición es de 1:25000, mucho menor que la reportada a nivel mundial.
La clínica es muy variable, desde síntomas leves, como somnolencia y fatiga, hasta vómitos, coma, convulsiones, rabdomiolisis, arritmias, cardiomiopatía, hepatomegalia, disfunción hepática y una clínica similar al Síndrome de Reye.8
Los hallazgos de laboratorio son: hipoglicemia no cetósica, elevación de transaminasas, hiperamonemia y acidosis láctica. Con frecuencia las crisis metabólicas son desencadenadas por periodos de ayuno prolongado, estrés, cirugías o infecciones.8
Se estima que aproximadamente un 1-3% de las muertes súbitas neonatales inexplicadas, obedecerían a un DOAG no diagnosticado de forma temprana. Sin embargo, con su incorporación en el Programa Nacional de Tamizaje en el 2006, la detección por medio de la espectrometría de masas en tándem, es fácil y rápida.9,10 En Costa Rica no se han documentado muertes súbitas neonatales relacionadas con un cribado positivo por DOAG.
Los DOAG presentan una herencia autosómica recesiva, por lo cual los padres son portadores de la enfermedad, con un riesgo de recurrencia del 25%. Debido a que existe la posibilidad de familiares asintomáticos con la deficiencia enzimática, es recomendable efectuar análisis bioquímicos y moleculares.
En cuanto al tratamiento de nuestro paciente, aun no existen medicamentos específicos para la DMD, pero las intervenciones médicas buscan disminuir las complicaciones y mejorar la calidad de vida. Los corticoesteroides presentan un potente efecto antinflamatorio, prolongan la deambulación al preservar la fuerza, disminuyen la progresión de la escoliosis y retardan el deterioro de la función cardiopulmonar. 4,11
El tratamiento debe ser valorado individualmente, tomando en consideración los efectos secundarios de los esteroides y la presencia de condiciones prexistentes en cada persona. Hay efectos adversos comunes a largo plazo, con la administración de corticoides, sobre todo en niños y en nuestro paciente en particular, por presentar otra enfermedad concomitante. Para un manejo exitoso, se debe valorar con regularidad el crecimiento, los niveles de glicemia y la presión arterial; es preciso reducir la dosis o cambiar el régimen de administración, en caso de que los efectos secundarios sean inmanejables.
Además, en este caso se debe proveer un control nutricional estricto, cuantificando la ingesta calórica y regulando los tiempos de alimentación, lo mismo que evitar los periodos de ayuno prolongado, con el fin de prevenir los episodios de hipoglicemias.
Asimismo, los hallazgos clínicos de fatiga, debilidad muscular y daño cardiaco, pueden ser observados en ambas condiciones, pero con tratamientos médicos diferentes, que los convierten en todo un reto médico para lograr el equilibrio necesario en su salud.
Conflicto de interés: ninguno
Referencias
2. Darras BT, Miller DT, Urion DK. Dystrophinopathies. Recuperado el 3 marzo de 2014. En: http://www.ncbi.nlm.nih.gov/books/NBK1119/
3. Matern D, Rinaldo P. Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. Recuperado el 3 marzo de 2014. En: http://www.ncbi.nlm.nih. gov/books/NBK1424/
4. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;1:77-93. [ Links ]
5. Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nature Genetics.1993;3:283-291. [ Links ]
6. Deconinck N, Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypothesis. Pediatr Neurol 2007; 36:1-7. [ Links ]
7. Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophinglicoprotein complex: An overview of current mouse models. Curr Opin Genetics Devel 2002;12:349-361. [ Links ]
8. Saudubray JM, van der Berghe G, Walter JH. Inborn Metabolic Disease. Diagnosis and Treatment. 5a ed.
9. Moczulski D, Majak I, Mamczur D. An overview of beta-oxidation disorders. Postepy Hig Med Dosw. 2009;63:266-277
10. Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33:521-526. [ Links ]
11. Merlini L, Cicognani A, Malaspina E, Gennari M, Gnudi S, Talim B et al. Early prednisone treatment in Duchenne muscular dystrophy. Muscle nerve 2003;27:222-227. [ Links ]
Afiliación de los autores: 1Pediatría,
2Microbiología-Genética Molecular. Hospital Nacional de Niños Dr. Carlos Sáenz Herrera *aleacosta_g@yahoo.com
Fuentes de apoyo: ninguna
Fecha recibido: 10 de diciembre de 2013 Fecha aceptado: 15 de mayo de 2014
{kind=link}