Linfohistiocitosis hemofagocítica, el espectro desde la enfermedad genética al síndrome de activación macrofágica
(Hemophagocityc Lymphohistiocytosis: A Spectrum from the Genetic Disorder to the Macrophage Activation Syndrome)
Servicio de Inmunología y Reumatología Pediátrica Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”
Abreviaturas: LHH, linfohistiocitosis hemofagocítica; LHHF, LHH familiar; cNK, células NK; TCD8, linfocitos T CD8+; SAM, síndrome de activación macrofágica; AIJs, artritis idiopática juvenil sistémica; CH, síndrome de Chediak-Higashi; SG, síndrome de Griscelli; XLP, síndrome linfoproliferativo ligado al cromosoma X; VEB, virus de Epstein-Barr; LHH-V, linfohistiocitosis hemofagocítica asociada a virus; LESj, lupus eritematoso sistémico juvenil.
Resumen
El compromiso de la regulación de la inflamación produce activación excesiva y expansión de macrófagos y linfocitos T que desencadenan una reacción inflamatoria severa, sin vías naturales de control. Los trastornos hemofagocíticos son la traducción clínica de este proceso inflamatorio. La linfohistiocitosis hemofagocítica se refiere a todas las variantes de esta patología, y el síndrome de activación macrofágica, a la variante asociada con enfermedad autoinmune. Los casos primarios se asocian con la forma familiar autosómica recesiva y los secundarios con inmunodeficiencias primarias, infección, malignidad y enfermedades autoinmunes. El principal distintivo de este grupo de patologías es la proliferación agresiva de macrófagos e histiocitos que fagocitan otras células sanguíneas. La reducción en la actividad de las células NK produce un aumento en la activación y expansión de linfocitos T, los cuales producen grandes cantidades de citoquinas. Las citoquinas inducen activación de macrófagos y células dendríticas, infiltración tisular y producción de interleuquinas, lo que genera una reacción inflamatoria severa, responsable del daño tisular y de las manifestaciones clínicas. El curso clínico se caracteriza principalmente por fiebre prolongada, hepatoesplenomegalia y citopenias. Los estudios de laboratorio muestran aumento de ferritina, triglicéridos e hipofibrinogenemia. La hemofagocitosis en médula ósea está presente en más del 80% de los casos al diagnóstico. El tratamiento está dirigido contra el linfocito T y los histiocitos hiperactivados, combinando quimioterapia con inmunosupresores y, en algunos casos, trasplante de células madre hematopoyéticas. Este tratamiento ha producido un cambio en la sobrevida de los pacientes. El protocolo de tratamiento HLH-2004 es una guía que estandariza el tratamiento, combinando etopósido, dexametazona y ciclosporina A. En Costa Rica se han reportado 60 casos en población pediátrica, con una mortalidad promedio del 44%.
Descriptores: linfohistiocitosis hemofagocítica, síndrome hemofagocítico, síndrome de activación macrofágica, lupus eritematoso sistémico juvenil, artritis idiopática juvenil sistémica, virus de Epstein-Barr, ferritina, hemofagocitosis, trasplante de médula ósea.
Abstract
Hemophagocytic lymphohistiocytosis is characterized by a severe hyperinflammatory condition, with macrophages and T cells activation and expansion, without regulatory pathways for the termination of the inflammatory activity. Hemophagocytic syndromes are clinical translation of an overwhelmed inflammatory response. The term hemophagocytic lymphohistiocytosis applies to all the variants of the syndrome and macrophage activation syndrome refers to the variant associated with autoimmune diseases. Primary cases are related to a familiar autosomic recessive disease and the secondary ones to primary immunodeficiencies, infection, malignancy and autoimmune diseases. Physiopathology of the disease is related mainly to the aggressive macrophage and histiocyte proliferation and phagocytosis of blood cells. An impaired function of NK cells and cytotoxic T-cells, increase cell activation and expansion and cytokine production inducing macrophage activation, tissue infiltration and tissue damage. The main symptoms are prolonged fever, cytopenias, hepatosplenomegaly, and hemophagocytosis. Biochemical markers include elevated ferritin and triglycerides and low fibrinogen. Bone marrow hemophagocytosis is present in more than 80% of the cases at diagnosis. Treatment targets activated T-cells an histiocytes, combining chemotherapy, immunosuppressors and in selected cases hematopoyetic stem cell transplantation. Treatment produced a positive change in patient survival. HLH-2004 treatment protocol is an standarized guideline that combine etoposide, dexamethasone and cyclosporine A. In Costa Rica 60 cases have been reported in children, with a 44% mortality.
Keywords: hemophagocytic lymphohistiocytosis, hemophagocytic syndrome, macrophage activation syndrome, juvenile-onset systemic lupus erythematosus, systemic juvenile idiopathic arthritis, Epstein-Barr virus, ferritin, hemophagocytosis, bone marrow transplantation.
La respuesta inflamatoria tiene mecanismos bien definidos para delimitar su duración y restablecer el equilibrio de la relación huésped-parásito, después de que el sistema inmune ha inducido inflamación. El compromiso de la regulación de la inflamación produce activación excesiva y expansión de macrófagos y linfocitos T que desencadenan una reacción inflamatoria severa, sin vías naturales de control. Los trastornos hemofagocíticos son la traducción clínica de este proceso inflamatorio y reflejan defectos que alteran la comunicación normal entre las respuestas del sistema inmune innato y adaptativo.1
En general, se ha relacionado esta patología con la edad pediátrica, probablemente por las descripciones iniciales de la forma familiar. Sin embargo, es un proceso inflamatorio que se puede presentar a cualquier edad, desde el recién nacido hasta el adulto mayor. Hay descripciones recientes del diagnóstico en adolescentes y adultos, de la forma familiar, en vinculación con cierto tipo de mutaciones.
El término correcto para llamar esta patología se ha prestado a confusión, en este artículo se usa linfohistiocitosis hemofagocítica, para referirse de manera inclusiva a todas las variantes del síndrome, y se deja síndrome de activación macrofágica, para indicar la variante asociada con enfermedades autoinmunes.
Clasificación
El término linfohistiocitosis hemofagocítica (LHH) fue acuñado por la “International Histiocyte Society” en 1998, para describir la LHH familiar (LHHF), una enfermedad genética con inflamación sistémica severa.2 Scott y Robb-Smith describieron el síndrome por primera vez en 1939, como reticulosis histiocítica medular.3 La descripción de LHHF es de Farquhar y Claireux, en 1952, quienes describieron la presentación pediátrica del cuadro con fiebre, hepatoesplenomegalia y citopenias.4 En el grupo de pacientes con enfermedades genéticas se incluyen los que tienen síndromes con inmunodeficiencia primaria.5
Los casos no familiares o adquiridos de LHH se describieron como secundarios y asociados con infecciones, malignidad, inmunosupresión y enfermedades autoinmunes. La estructura de clasificación, basada en las recomendaciones de la “Histiocyte Society” para las patologías del histiocito, se muestra en el Cuadro 1.6 En ocasiones, en publicaciones de casos en adultos se utiliza el término “síndrome de activación macrofágico reactivo”, para indicar LHH adquirida o síndrome hemofagocítico, sin tomar en cuenta la patología de fondo.7

El principal distintivo de este grupo de patologías es la proliferación agresiva de macrófagos e histiocitos que fagocitan otras células sanguíneas. Es un crecimiento de células descontrolado, sin características de malignidad y sin clonalidad, con migración ectópica, principalmente a bazo, ganglios linfáticos, médula ósea, hígado, piel y las membranas que cubren la médula espinal.
La teoría actual que explica la fisiopatología de esta enfermedad, identifica una reacción inmunológica anormal, como consecuencia de la activación de linfocitos T y de macrófagos, sin la intervención de los mecanismos de apoptosis normales, que conduce a inflamación sistémica. Es un problema de función más que de ausencia de estas células; la infiltración tisular puede producir una linfopenia TCD8+ paradójica. Una respuesta Th1 descontrolada y una función citotóxica defectuosa son los mecanismos fundamentales en la fisiopatología de este grupo de patologías.8-10
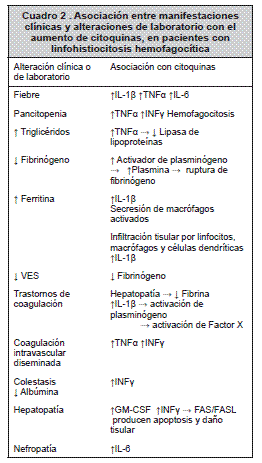
Se ha identificado una deficiencia en la función citotóxica de células NK (cNK) y linfocitos T CD8+ (TCD8). En el mecanismo normal de citotoxicidad, las cNK y los TCD8 destruyen sus células blanco utilizando una vía no secretora (FasL, CD95L) y una secretora, liberando gránulos citotóxicos que contienen perforinas, granzima y otras sustancias proteolíticas. La célula citotóxica y la célula blanco forman una sinapsis inmunológica en el punto de contacto, hacia el cual rota el centro de organización de microtúbulos, se liberan los gránulos citotóxicos que se fusionan con la membrana y liberan su contenido en la sinapsis, se producen poros en las membranas celulares y, como consecuencia, lisis osmótica, degradación proteica y apoptosis. 5, 11 La reducción en la actividad de cNK produce un aumento en la activación y expansión de linfocitos T, los cuales generan grandes cantidades de citoquinas (interferón gamma, factor de necrosis tumoral alfa, factor estimulante de colonias de granulocitos-macrófagos). Las citoquinas inducen activación de macrófagos y células dendríticas, infiltración tisular y producción de interleuquinas (IL-6, IL-2, IL-1β, IL-8, IL-10, IL-12, IL-18), lo que produce una reacción inflamatoria severa, responsable del daño tisular y de las manifestaciones clínicas. Cuando se determinan niveles de citoquinas en suero se encuentran concentraciones de 10 a 1000 veces mayores que las normales. 5, 10, 12-14
Los gatillos que disparan esta serie de eventos probablemente utilizan vías propias de cada uno de ellos (virus, bacterias, hongos, autoinmunidad, medicamentos). En los pacientes, durante la etapa activa de la enfermedad, se detectanniveleselevadosdevariascitoquinasproinflamatorias, las cuales producen alteraciones progresivas de diferentes órganos, que causan a la muerte del paciente. 15
Además, se puede presentar pérdida rápida de peso, sangrado por alteración de los tiempos de coagulación (prolongación de TP y TPT), infiltrados pulmonares y compromiso cardiaco y renal.5, 9, 10
La hematología muestra anemia arregenerativa y trombocitopenia de aparición temprana; la mitad de los casos tienen neutropenia, y la leucopenia es menos frecuente y más tardía en el curso de la enfermedad. Tres de cada cuatro pacientes sufren pancitopenia y todos, bicitopenia. En otros estudios de laboratorio se identifica un aumento de ferritina, triglicéridos, transaminasas, bilirrubinas y deshidrogenasa láctica e hipofibrinogenemia, que provoca disminución en la VES. El trastorno de la coagulación más frecuenteesladeficienciaaisladadefibrina,comoconsecuencia de alteraciones en la función hepática y por la activación por IL-1β de plasminógeno y de factor X. Aunque no es frecuente, pero sí asociada con alta mortalidad, se puede presentar coagulación intravascular diseminada por la sobreproducción de interferón gamma y de factor de necrosis tumoral alfa.10, 15
Citolisis y colestasis están asociadas con alteración de la función hepática. Interferón gama favorece el desarrollo de colestasis e hipoalbuminemia. La interacción Fas/Fas ligada en respuesta a la sobreproducción de interferón gama, explica la apoptosis y el daño del tejido hepático.
Se pueden detectar otras alteraciones asociadas con el proceso inflamatorio, como hipo o hipergammaglobulinemia, Coombs directo positivo e hiponatremia asociada a secreción inapropiada de hormona antidiurética.1
Algunas de las manifestaciones clínicas se pueden interpretar como la respuesta inflamatoria normal hacia un agente infeccioso, pero la severidad indica progresión y falta de respuesta al tratamiento debe hacer sospechar LHH.
El estudio del aspirado de médula ósea muestra evidencia de eritropoyesis inefectiva, con incremento de la eritropoyesis, pero elevación moderada de reticulocitos; la actividad prolongada del proceso inflamatorio puede producir una médula ósea aplásica. La hemofagocitosis en médula ósea está presente en más del 80% de los casos al diagnóstico, pero puede estar ausente al inicio del cuadro inflamatorio.10
 ]]>
]]>
La mortalidad de LHH se reporta entre un 22 y un 59%. Las variantes asociadas con malignidad o infección por VEB son las que tienen las tasas de mortalidad más altas. En los casos que fallecen, la muerte ocurre durante las primeras 4 a 8 semanas y está asociada con falla orgánica multisistémica, sangrado o sepsis.10

