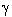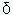Síndromes talamésicos. Nuevos conceptos y estado actual del conocimiento en Costa Rica
German F.Sáenz-Renauld. Walter Rodríguez-Romero
Resumen
En tanto que para las hemoglobinas Hbs anormales es posible determinar los lugares de su origen (marcadores antropológicos), en las enfermedades talasémicas se tiene el problema de su amplia distribución en las principales poblaciones ancestrales. Las talasemias resultan ser las enfermedades monogénicas más comunes en el mundo. La amplia dispersión de la talasemia en todo el Mediterráneo, el Medio Este de Asia, el sudeste Asiático, el subcontinente Indio y otras regiones, indica que sus orígenes genéticos fueron independientes. De igual manera acontece con las alfa talasemias (sudeste asiático, el sur de China, las Filipinas, África y el Mediterráneo). En Costa Rica la alfa talasemia se encuentra en raza negra, con los dos alelos (23% para el +:(1)y 3.9 para el º (2); y han sido esporádicos los casos doble heterocigotos que originan la enfermedad por Hb H (+/º), siempre en individuos de extracción oriental. Especial mención se hace de la talasemia menor y de su importante diferenciación con la anemia ferropriva. Al presente se han descrito unos 8 casos de talasemia mayor en el país, y no son infrecuentes las formas clínicas intermedias de esta talasemia con o sin Hb S. Un breve comentario hace ver algunos aspectos novedosos de la fisiopatología de la talasemia y su tratamiento en los países en desarrollo.
Descriptores: Hbs, Hemoglobinas A, A2 y F; Hemopoyesis, genes de globina y ; mutaciones puntuales; delecciones; deficiencias génicas; microcitosis hipocrómica; ferropenia; beta talasemia menor, mayor e intermedia; enfermedad heterocigota al talasemia y Hb H; ferritina; hemicromos; fosfatidilserina; apoptosis; malaria.
Key words: Hemapolesis, genes of globulin and ; hemoglobin A, thalassemia.
Recibido:6 de marzo de 2006 Aceptado:18 de julio de 2006
Todas las hemoglobinas (Hb) humanas consisten de dos diferentes pares de cadenas de globina combinándose cada una con la ferroprotoporfirina (hem). La Hb F se sintetiza durante la vida fetal y declina después del nacimiento. Posee dos cadenas alfa y dos gama. En el adulto, la principal Hb es la A que posee dos cadenas alfa y dos beta. Un menor componente lo constituye la Hb A2 que posee dos cadenas alfa y dos delta. Los genes alfa y gama se hallan duplicados en el cromosoma 16, en tanto solo se presenta un gene por genoma haploide en el cromosoma 111,2. Las talasemias son las más comunes enfermedades monogénicas en los humanos. Aproximadamente un 1.7%, de la población mundial es heterocigoto para alfa y beta talasemias3. Estos trastornos hereditarios de la Hb se caracterizan por una reducida síntesis de una de las cadenas de globina de la Hb adulta (Hb A), constituyendo un problema de salud pública en lugares donde estos trastornos son endémicos, como sucede en la Europa Mediterránea, para el caso de las beta talasemias.
]]>
Las talasemias son el resultado de mutaciones o pérdidas de genes, habiéndose demostrado más de 200 diferentes mutaciones puntuales en los genes beta y alfa de la globina, y rara vez lo son por delección3. Las talasemias se clasifican de acuerdo con la cadena de globina que se produce en forma ineficaz. Con mucho, el alfa y la betatalasemia son las más importantes, aunque también existen los genotipos de delta/beta y gama/delta/betatalasemia2. En muchas poblaciones en las cuales la talasemia es frecuente, también pueden ser comunes los genes para Hbs estructurales (S, C, E), por lo que no es inusual que individuos de aquellas hereden ambos genes, el de la talasemia y el de la mutante hemoglobínica. Los genotipos más comunes que surgen de estos casos doble heterocigotos lo constituyen la Hb S/betatalasemia y la Hb E/beta-talasemia. La primera condición se observa en nuestro país, especialmente en las poblaciones de los litorales4-6.
Las beta talasemias:Debido a su conexión con niños de la región mediterránea, la enfermedad se denominó talasemia, derivada de la palabra griega talaza que significa mar. Las talasemias se encuentran dispersas por toda la región del Mar Mediterráneo, África del Norte-ecuatorial, el Medio Este de Asia, el subcontinente Indio, Burma, el sudeste asiático, incluyendo la península de Malaya e Indonesia, y sus orígenes son independientes. Se ha postulado, al igual de como se ha establecido para otras hemoglobinopatías y eritroenzimopatías, que estos "genes" talasémicos presentan una ventaja adaptativa en los heterocigotos a la infección por P. falciparum7. Las beta talasemias pueden ser de dos tipos: las beta+-talasemia +, en las cuales el trastorno mutacional da como resultado una síntesis defectuosa de cadenas beta, y las betaº-talasemias º, en las cuales no se produce cadena beta, por lo quel se le conoce como beta talasemia supresora. En la condición heterocigota o talasemia menor (sea + ó º), el desbalance biosintético de la síntesis de cadenas de globina origina un exceso de cadenas alfa, con la correspondiente hipocromía eritrocítica y ligera afectación de la sobrevida del eritroblasto y del eritrocito circulante. En el hemograma de estas formas menores se destaca una eritrocitemia microcítica hipocrómica y una elevación de la Hb A21,2, con una disminución de 1 a 3 g/dI de la Hb total según edad y sexo8. Si en vez de la Hb A2 es la Hb F la fracción menor que predomina, se denomina delta/beta-talasemia9. En el caso homocigota o mayor de beta talasemia (enfermedad de Cooley), el exceso de cadenas alfa libres (ante la marcada disminución o ausencia de cadenas beta) precipita en los eritroblastos y su sobrevida se toma crítica; se produce una severa anemia, la cual en turno lleva a un incremento en la producción de eritropoyétina y a una expansión de una medula ósea ineficaz, con deformidad ósea, esplenomegalia y retardo en el crecimiento (Cuadro 1). Hay un aumento en la síntesis de la Hb F que no compensa el defecto severo o la ausencia total de síntesis de Hb A. En la forma homocigoto de beta+-talasemia y en la doble heterocigota betaºtalasemia/ beta+-talasemia, la clínica es parecida a la que se observa en la +, aunque menos severa por el hecho de que hay cierto grado de síntesis de Hb A (< de 30%). Algunos de estos casos podrían caber en lo que se denomina talasemia intermedia10, cuadros hematológicos que pueden no llegar a ser transfusión dependientes. En el Cuadro 1 se pueden apreciar diferentes determinantes fenotípicos de talasemia, la mayoría de ellas halladas en el país.
Generalidades clínicas y de laboratorio de la talasemia:La clásica talasemia menor puede pasar inadvertida por años, o verse por mucho tiempo como si fuera anemia por deficiencia de hierro, debido a las similitudes hematológicas entre ambas condiciones, y por ser la ferropenia más frecuente que las talasemias. Por otra parte, una deficiencia concomitante de hierro puede deprimir los niveles de la Hb A2 en talasemia menor2, hecho que puede establecer una confusión diagnóstica importante, especialmente en mujeres embarazadas y en la niñez temprana. Lo primordial es detectar a los individuos portadores de talasemia menor para descartar la posibilidad de que la progenie herede la forma homocigota de la enfermedad.
La gestación en mujeres con talasemia menor se complica primordialmente por un aumento en la gravedad de la anemia en el lapso de las 24 a 28 semanas de gestación. En un total de 308 pacientes recopilados de la bibliografía médica, el nivel mínimo medio de hemoglobina fue de 9.3 g/dI8. Esto es ciertamente compatible con una gestación normal y, en la mayoría de casos, no fue necesaria ninguna otra terapéutica. Aunque el diagnóstico bioquímico de laboratorio está bien establecido, existen talasemias silenciosas1, que requieren un estudio genético-molecular, como también para las formas mixtas de y talasemia, o cuando se suscita la coexistencia de un determinante talasémico con otra condición anémica (ej., deficiencias de hierro, folátos, vitamina B12, esferocitosis, etc).
La talasemia mayor es una enfermedad severa. Se diagnostica usualmente entre los 6 y 12 meses de vida, en niños que nacieron con valores de hemoglobina normales y que con el transcurrir del tiempo comienzan a sufrir de un cuadro anémico progresivo. Ello se explica por la disminución paulatina de los niveles de la Hb fetal. Los niños con la condición mayor cursan con anemia de 4 ó 5 gramos de Hb, presentan hepato-esplenomegalia y una anemia hemolítica marcada. Estos pacientes por lo general son dependientes vitalicios de transfusiones sanguíneas, si no se siguen otras vías de tratamiento.
La hemolisis y la eritropoyesis ineficaz son los dos mecanismos de anemia en los síndromes talasémicos mayores. La ineficacia de la síntesis de Hb es producto de una acelerada apoptosis causada por un exceso de cadenas alfa depositadas en los precursores eritroides. Esta apoptosis incrementada se asocia con un aumento de la expresión de la fosfatidilserina, una importante señal para la remoción prematura por macrófagos medulares hiperplasiados de eritroblastos con regidez de su citoesqueleto3. En las formas mayores de beta talasemia los cuadros fisiopatológicos de hemolisis y de hipercoagulabilidad se deben a la oxidación de las subunidades , ó de la Hb, lo cual lleva a la constitución de hemicromos cuyo grado (mayor en las cadenasa) determina el grado de hemolisis3. Estos hemicromos se unen o modifican a varios componentes de la membrana eritrocitica. Luego de su precipitación como cuerpos de inclusión, el hem se desintegra y se libera hierro que cataliza la formación de especies de oxigeno reactivo, las cuales, entre otros daños celulares, propician la generación aumentada y exteriorizada de fosfatidilserina lo que provoca una activación del sistema de la coagulación y de las plaquetas11, a semejanza de lo que ocurre en la drepanocitosis12.
Los regímenes regulares de transfusión sanguínea pretenden mantener niveles de Hb entre 9 y 10 g/dl, con el fin de mejorar el crecimiento y el desarrollo, reducir la hepatoesplenomegalia y las deformaciones óseas. La sobrecarga de hierro causa la mayor parte de la morbilidad y mortalidad asociadas con la talasemia. El exagerado depósito de hierro ocurre en órganos viscerales (en especial corazón, hígado y glándulas endocrinas), lo que causa daño tisular, disfunción y fallo orgánico, siendo la sobrecarga de hierro cardíaco la causa primaria de muerte3. La clásica deferoxamina es un útil agente quelante de hierro, pero tiene sus limitaciones clínicas y de costo13. El moderno desarrollo de quelantes activos por vía oral ha significado un valioso avance para el tratamiento de la hemosiderosis de la talasemia. Dentro de ellos se citan la deferiprone14 y el deferasirox(ICL 670)15.
Un aumento en la síntesis de la Hb F puede mejorar la severidad de la talasemia. Para lograr este propósito se han usado varios productos, entre ellos la hidroxi-urea16, con un éxito esperanzador inicial que requiere mayor confirmación. De igual manera se halla lo relativo a los transplantes de médula ósea no ablativos con el uso de células madres medulares o de cordón umbilical3. El buen resultado que se obtiene con el clásico transplante medular17, permite a un número apreciable de pacientes el abandono de la terapia, las transfusiones y la quelación de hierro3. En algunas regiones del Mediterráneo donde los portadores de talasemia menor alcanzan más del 3%, se tienen programas de trasplante de médula ósea2, los cuales se aconseja practicar en la niñez temprana, antes de que se hayan desarrollado las complicaciones provocadas por la sobrecarga de hierro1. El tratamiento molecular experimental se basa en un enfoque directo sobre el gene talasémico de beta globina, a través de inductores ya sea de Hb F, o por la inserción de genes funcionales de globina gama o de globina beta. El uso de ratones talasémicos transgénicos está dando la oportunidad de lograr en un breve plazo algún tratamiento adecuado para la talasemia en humanos18,20, tal y como está siendo logrado con modelos de ratones sicklémicos transgénicos, en los cuales se está obteniendo una corrección de la anemia drepanocitica inducida21.
]]>
En Costa Rica se han encontrado al menos ocho casos de talasemia mayor. El primero se reportó en 1976 con dos posibles genotipos: º/+ ó +/+-talasemia18. En 1981 se reportó el segundo hallazgo, también con genotipo compatible, como en el caso anterior. En ambas comunicaciones se hizo evidente la presencia de al menos un alelo +-talasemia, por la existencia de Hb A en el hemoglobinograma de los proposita. En el tercero se reporta una -talasemia mayor o intermedia10, en la que se logró demostrar un genotipo º/()º, con una repercusión clínica atenuada del trastorno, en virtud de la moderada compensación dada por los niveles de incrementados de la Hb fetal, a pesar de la ausencia de Hb A. La cuarta enfermedad talasémica homocigota fue de tipo º/º-talasemia19, con total ausencia de Hb A y, como era de esperarse, con severa repercusión clínica, constituyéndose en el primer caso nacional en donde concurren dos alelos talasémicos de carácter supresor (bo-talasemia). Otros cuatro casos se han estudiado en los últimos años con genotipo variable, y de nuevo con presencia del gene º asociado al frecuente codón (C-T 39). Precisamente esta mutación sin sentido es la que predomina (63% de los casos) en los estudios que realiza el CIHATA en nuestro país.
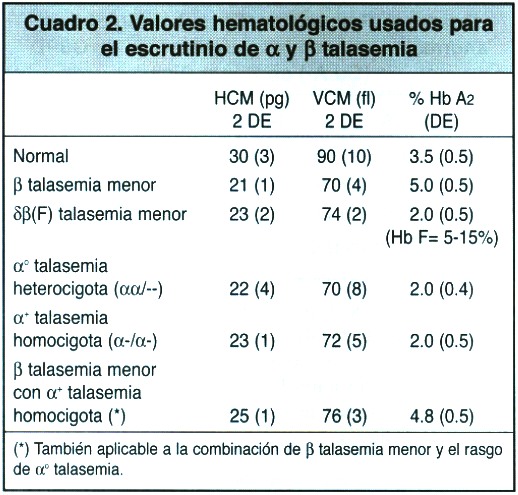
Las alfa talasemias:Las alfa talasemias muestran varias diferencias importantes respecto de la beta talasemia. En vista de que las cadenas alfa forman parte tanto de la Hb F como de la Hb A, algunos síndromes de alfa talasemia se manifiestan ya en la edad fetal. Como los genes alfa se hallan duplicados en el genoma respectivo, la genética de las alfa talasemias es más complicada que la de las beta talasemias (Cuadro 2). El genotipo normal, por lo tanto, se designa -/-. La pérdida de un gene se designa + talasemia (/-) y corresponde a la forma silenciosa; la pérdida de dos corresponde con la º tal (-/-) o forma heterocigota.
La pérdida de un gene en cada cromosoma origina un genotipo idéntico de º (-/-; también conocida como condición homocigota de + talasemia). La ausencia total de genes (--/--)es incompatible con la vida y originar un tetrámero inestable 4 (que provoca hydrops fetalis de Bart). La llamada enfermedad por Hb H (4) es una condición doble heterocigota de + talasemia y de º talasemia (--/-). Tanto el portador de + talasemia como el de º talasemia se han descrito en el país, siendo preponderantes en la raza negra 20. La enfermedad por Hb H solo se ha demostrado en población oriental que radica en el país 21,22. La a talasemia ocurre en muchos grupos étnicos, siendo especialmente común en chinos, en el Sudeste Asiático y en el África Occidental, como también en el Mediano Este y en el Área Mediterránea. En asiáticos los haplotipos de talasemia que predominan son el --/(º)y el clásico -/(+), cuya concurrencia alélica origina la enfermedad por Hb H.
En raza negra predominan el +, siendo menos frecuente el fenotipo º, el cual, por otra parte, es genotípicamente producto de la herencia dual del +, por lo que el genomadiploide del carácter determinante de º talasemia corresponde en esa raza al genotipo -/-y no al --/de los asiáticos1,2,23. De esta forma, en raza negra el rasgo o forma heterocigota de a talasemia (º) se debe fundamentalmente a una homocigocidad para el gene + talasémico, lo que explica el hecho de que la enfermedad por Hb H (--/-)sea excepcional en esa raza y nunca se haya observado la forma letal de º homocigota1,2. En 280 muestras de cordón umbilical de niños recién nacidos de raza negra de la provincia de Limón20, se pudo ]]>
+ talasemia, con una cifra del 3.9% para el gene severo o heterocigota (
º), de acuerdo con los valores de Hb Bart (3-8%). La experiencia en el CIHATA, indica que la principal delección (26% de los casos) de a talasemia en raza negra costarricense es por la pérdida de 3.7 kb, y que un 20% lo sea por la delección -SEA (sudeste asiático) en personas de origen oriental. Los portadores de º talasemia (--/)y los homocigotos para + ]]>
-/-)presentan un cuadro hematológico de anemia microcitica hipocrómica que recuerda a la clásica talasemia menor (Cuadro 2). El portador silencioso de + talasemia (-/)no muestra ninguna alteración en los índices hematológicos. Usualmente el corte de valores para el VCM de 70 fl y para la HCM de < 25 pg se usa para indicar la necesidad de estudiar niveles de Hb A2 y/o Hb F, sin embargo, en poblaciones en las cuales son frecuentes la y talasemias, aquellos índices eritrocíticos se deben elevar a 80 fl y 27 pg.
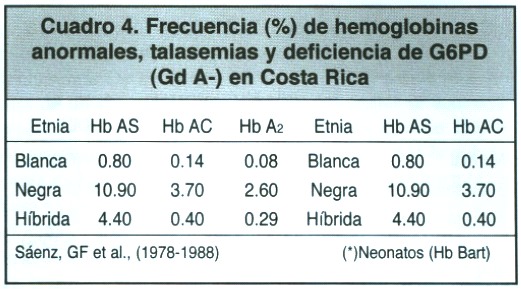
Experiencia nacional en torno a las talasemias:La primera evidencia sobre la presencia de la beta talasemia en nuestros países se halla registrada en el libro de Hematología del Dr. Wintrobe (2ª. Edición, 1961), quien personalmente estudió una familia costarricense de ascendencia española y escocesa. Es lógico pensar que los casos de talasemia encontrados en Costa Rica sean producto de la inmigración de diversos individuos de países en donde estos defectos hereditarios de la Hb son frecuentes (Cuadro 3). Se podría aceptar que nuestros aborígenes, al igual que los de otros países latinoamericanos, no poseen genes de talasemia y que las pocas comunicaciones -fuera de Costa Rica- en que se ha citado su presencia sean producto de entrecruzamientos relativamente recientes, tal y como se ha explicado para la presencia de Hb S en aquella etnia24. En la época actual, al romperse las cadenas sociales y asimilarse la etnodilución por las variadas y accesibles comunicaciones del tránsito humano, la situación podría variar ostensiblemente. Dada la marcada ascendencia española de nuestras poblaciones, se cita la frecuencia de -talasemia en España, la cual se halla en el ámbito del 1.10% al 3.43%, en tanto que la -talasemia parece ser mucho menos frecuente. También se encuentran amplias variaciones en la frecuencia de ambos trastornos talasémicos en África y en países mediterráneos. Por otra parte, es imposible, la mayor parte de las veces, establecer el origen étnico de un gene talasémico específico, ya que el fenotipo clínico de cada forma de talasemia es similar en todos los grupos étnicos. Por otra parte, la -talasemia usualmente moderada en raza negra, en ocasiones muestra el mismo cuadro clínico que se observa en europeos.
]]>
En la bibliografía nacional, el primer informe sobre la presencia de un gene de -talasemia del tipo + en conjunto con Hb S, lo brindaron Zomer y Rivera en 19674. Posteriormente, en 1973, Zomer et al25 evidencian 11 casos de talasemia menor tipo Hb A2alta en una muestra hospitalaria seleccionada. En 1975, Sáenz et al9 demuestran la existencia de -talasemia o F-talasemia en población universitaria caucásica y caucasoide. Este tipo de F-talasemia se caracteriza por un aumento de la Hb F con valores que oscilan entre el 5-15%, y niveles normales o disminuidos de la Hb A2. El gene º (supresor) se destaca en una publicación de Sáenz et al5 en un paciente doble heterocigoto S/º talasemia, con importante repercusión clínica semejante a la drepanocitosis.
La prevalencia global de beta talasemia menor en Costa Rica está bien documentada. Se destaca en uno de los trabajos nacionales el hallazgo de una frecuencia del 0.25% del rasgo, en 12.000 niños de una población escolar representativa de todo el país 26 del 0.70% en raza negroide27 del Guanacaste, y del 0.4% en raza negra de Limón (Cuadro 4).
La necesaria distinción entre talasemia menor y anemiaferropriva:El diagnóstico de beta-talasemia menor pasa a menudo inadvertido por muchos años, siendo muy importante establecer el diagnóstico lo más temprano posible, para que el paciente deje de sufrir continuas investigaciones y tratamientos medicados. Por lo general, la talasemia menor es un diagnóstico de laboratorio, pues no puede ser sospechada solo con las evidencias clínicas, al tratarse de trastornos que básicamente, y salvo complicaciones, son asintomáticos, al cursar con un nivel de Hb de 1 a 2 grados por debajo del límite inferior por edad, sexo y altitud2. Estos pacientes pueden pasar inadvertidos y diagnosticarse, por lo tanto, en edad adulta.
El problema práctico más común en los estudios de talasemia es distinguir la -talasemia menor (sea bº,b+,o ) de la anemia por deficiencia de hierro. Esta discriminación puede originar problemas diagnósticos. Por supuesto, si se conocen los valores de hierro sérico o de ferritina, y es asequible una estimación de la Hb A2, ello ayudaría a discriminar ambas condiciones. Sin embargo, los parámetros citados arriba no son rutina en todos los laboratorios. La protoporfirina eritrocítica libre o la ligada al Zinc (PP-Zn), se halla incrementada en la deficiencia de hierro, pero no en el rasgo de -talasemia, en la mayoría de los casos. La medición de ferritina o de protoporfirina en escrutinio de grandes poblaciones puede ser muy costosa y lenta, y por lo tanto no sería práctica. En las comunidades en donde ambas alteraciones son frecuentes, puede haber mayores dificultades diagnósticas, especialmente cuando existe una deficiencia severa de hierro más -talasemia, situación frecuente en la mujer embarazada y en la niñez temprana. En estos casos, los niveles de la Hb A2 pueden disminuir y solamente se restauran luego de la terapéutica con hierro. A un nivel de Hb no menor de 10 g/dI, la deficiencia de hierro usualmente no se asocia con una obvia microcitosis e hipocromía, hecho que sí es característico en los rasgos de -talasemia y de -talasemia heterocigota (º).Por lo tanto, una regla útil es que cuando se detecta microcitosis e hipocromía (observable ya sea en el frotis sanguíneo o por medio de contadores electrónicos), asociados a niveles de Hb de 10 g/dl o mayores, ello podría significar más un rasgo talasémico que una deficiencia de hierro.
Otras dificultades diagnósticas pueden ocurrir cuando coexiste con la -talasemia clásica la condición heterocigoto para º + talasemia. Por otro lado, existen dos formas o tipos de talasemia menor con niveles normales de la Hb A2: el cuadro ligero silencioso (tipo 1), en donde a lo sumo se aprecian ligeros cambios eritrocíticos, aunque la relación sintética /de 1:3 es del tipo clásico de -talasemia, y el tipo silencioso 2, o severo de -talasemia menor, en el cual sí existen cambios morfológicos característicos. La herencia homocigota con cualesquiera de estos determinantes da como resultado cuadros clínicos mayores de talasemia.
Con el advenimiento de los modernos equipos automatizados de hematología que utilizan para la citometría cuantitativa la impedancia electrónica, se logra determinar con gran precisión, entre otros parámetros, el tamaño de los eritrocitos, se ha podido demostrar que los eritrocitos de los sujetos talasémicos son de un diámetro más uniforme que los eritrocitos anormales, microcíticos hipocrómicos de los pacientes con deficiencia de hierro. Con los equipos aludidos es posible integrar histogramas de la distribución del volumen de los eritrocitos; medido como coeficiente de variación, se informa como anchura de la distribución de los eritrocitos o RDW (siglas de su designación en inglés: "red cell distribution width"). Este índice es el equivalente de la anisocitosis, pero con la ventaja de ser independiente de la variable apreciación personal del observador y cuyos valores de referencia oscilan entre el 11.6% y el 14.8%. Se aplica para distinguir presuntamente las talasemias con RDW dentro de los límites normales de referencia o ligeramente elevados- de las anemias por deficiencia de hierro que originan valores superiores. Esta orientación y la medición en sangre de hierro sérico, capacidad de fijación de hierro, índice de saturación de la transferrina, ferritina en suero y protoporftrina de los eritrocitos, permiten distinguir ambas posibilidades diagnósticos. El seguimiento de estas orientaciones posibilita establecer el diagnóstico en la mayor parte de los casos y es deseable que los clínicos las apliquen en la práctica. Sin embargo, hay evidencia de que están siendo subutilizadas, pues muchos médicos no conceden importancia a la información que los laboratorios proporcionan, como lo demuestra un estudio realizado en un hospital de enseñanza estadounidense28, en donde se refiere la inadecuada interpretación que varios médicos conceden a la información suministrada por los laboratorios de hematología:
]]>
1. Las anormalidades de los índices eritrocíticos fueron advertidos, por médicos no hematólogos, únicamente en un 50% de los pacientes.
2. En el 68% de los casos con índices compatibles con talasemia, esta posibilidad de diagnóstico no se consideró.
3. En los pacientes en quienes la talasemia se incluyó en el diagnóstico diferencial y se investigaron anormalidades de la hemoglobina, solo el 56% de los estudios se interpretaron correctamente.
4. En el 17% de los casos los pacientes se trataron con hierro, sin que los exámenes de laboratorio confirmaran la deficiencia.
5. En los enfermos, a pesar de que sus estudios sugerían síndrome talasémico, se estableció el diagnóstico de deficiencia de hierro con base en la existencia de microcitosis y se les administró hierro innecesariamente, aún en aquellos que tenían parámetros normales de hierro.
Finalmente, caben otras observaciones: en todos los pacientes con microcitosis e hipocromía deben estimarse parámetros de hierro para ratificar o descartar su deficiencia. No debe prescribirse ferroterapia, particularmente en su forma parenteral, sin demostrar deficiencia de hierro. Si en las personas con microcitosis e hipocromía no se demuestra deficiencia de hierro, debe cuantificarse la Hb A2. Si se encuentra elevada, se establece el diagnóstico de talasemia heterocigota. Asimismo, si la Hb A2 se halla dentro de límites normales, es probable que el paciente sea portador de talasemia silenciosa con Hb A2 normal, de talasemia º ó de talasemia (con la Hb F elevada). En ambos casos está indicado practicar el análisis de la síntesis de cadenas y los estudios pertinentes de biología molecular y de una electroforesis inicial con cuantificación de la Hb F. Como no existe tratamiento alguno para estas anormalidades, el médico debe convencer al paciente de que cualquier tratamiento es innecesario, insistiendo en que la administración de hierro, particularmente de sus formas parenterales, además de ineficaz, puede ser dañina. Deben discutirse las implicaciones genéticas con estas personas y, en caso necesario, estudiar a otros miembros de la familia para descubrir portadores adicionales. El consejo genético entre parejas portadoras se orientará a la profilaxis de descendientes homocigotos.
Rund y Ranchmilewitz3,en torno al tratamiento de la talasemia mayor, expresan el siguiente comentario: En las partes desarrolladas del mundo, como los Estados Unidos y Europa, hay aproximadamente 10.000 pacientes homocigotos con talasemia y la cantidad de casos nuevos ha ido disminuyendo progresivamente debido a los métodos eficaces de prevención. En estos países está disponible la atención médica comprensiva y de alta calidad, con una mayor expectativa de vida y una calidad de vida relativamente buena. Los países occidentales enfatizan la terapia curativa, como el transporte de médula ósea y la terapia genética, que requieren que los pacientes cumplan las indicaciones y tengan acceso a los medicamentos más novedosos e instalaciones científicas sofisticadas. Las culturas occidentales deben desarrollar un mejor apoyo para los pacientes con talasemia y sus familias, ya que con esto pueden prevenir los aspectos psicosociales importantes que tienen tanto peso por la falta de acatamientos de las indicaciones.
En contraste, el asunto de la talasemia es totalmente distinto en los países menos desarrollados, en donde vive la mayor parte de los pacientes con esta enfermedad. No están disponibles universalmente las transfusiones seguras (con el uso de filtración y pruebas virales de la sangre) y la quelación, por lo que muchos pacientes con talasemia en los países subdesarrollados mueren en la infancia o la adolescencia. Deben establecerse programas que brinden una atención aceptable, que incluya la transfusión de sangre segura y la terapia de apoyo, como la quelación. En estos países, es preciso desarrollar protocolos que incluyan una mejor educación y tamizaje, así como un más adecuado acceso a los procedimientos diagnósticos prenatales. El reto para el futuro reside en asegurar que las personas que nazcan con una forma grave de talasemia sigan prosperando, y que la prevención eventual reduzca en todo el mundo la cantidad de pacientes gravemente afectados.
Abstract
While for abnormal hemoglobins it is possible to use anthropological markers to establish their origins, the thalassemias have a very wide distribution in ancestral populations. Thalassemias are the world's most common monogenic disorders. The dispersion of thalassemia in the Mediterranean region, the Middle East, the southeastern part of Asia, the Indian subcontinent and other regions, indicate that their genetic origins were independent.
]]>
thalassemia shows a similar pattern (southeastern part of Asia, southern China, Philippines, Africa and the Mediterranean). In Costa Rica thalassemia is found in the black population with two alleles (+ (1) 23% and º (2) 3.9%). Sporadic double heterozygous cases of Hb H disease (+/º), are from oriental population. Special reference is made of minor thalassemia and their important differentiation with iron deficiency anemia. Eight major thalassemia cases have been described in our country. Intermediate thalassemia cases with or without Hb S are not infrequent. A short commentary is made of the pathophysiology of thalassemia and the treatment in developed countries.
2. Weatherall DJ. The thalassaemias (Forthigty review). Brit Med J, 1997; 314: 1675-1679. [ Links ]
3. Rund DP; Rachmilewitz E. -thalassemia (Review article; medical progress). N Engl J Med, 2005; 353:1135-46.
4. Zomer M; Rivera A. Primer caso de hemoglobinopatía S/talasemia en Costa Rica. Acta Med Cost. 1967; 10: 71-74. [ Links ]
5. Sáenz GF;Elizondo J;Páez CA. Hallazgo del gene + talasémico (supresor) en Costa Rica. V: Síndrome de heterocigosis doble (S/betaº-tal). Sangre, 1978; 2: 192-204.
6. Sáenz GF;Sánchez G; Monge B. Síndromes drepanocíticos en Costa Rica. IV. Hemoglobina S/delta-beta talasemia (S/Ftal). Acta Med Cost. 1976; 4: 3-9. [ Links ]
7. Weatherall DJ. Thalassaemia and malaria, revisseted. Ann Trop Med Parasitol. 1997; 91: 885-890. [ Links ]
8. Necheles T. Complicaciones obstétricas asociadas con hemoglobinopatías. En: Clínica Hematológica, 1974; 66-83, Salvat Ed. S.A., Barcelona. [ Links ]
9. Sáenz GF;Alvarado MA; Arroyo G. F (delta-beta) talasemia en Costa Rica. Acta Med Cost. 1974; 1: 63-66. [ Links ]
10. Sáenz GF;Carrillo JM;Mora L; Chaves M; Jiménez R. Beta talasemia intermedia de genotipo º/(c)o. Comunicación de un caso. Sangre, 1984; 29: 467-472.
11. Eldor A; Rachmilewitz FA. The hypercoagulable state in thalassemia. Blood 2002; 99:34-43. [ Links ]
12. Znaal RF; Schrort AJ. Pathophysiology implications of membrane phospholipid asymmetry in blood cell. Blood 1997; 89:1121-1132. [ Links ]
13. Oliviere NF. The -thalassemias. N Engl JMed. 1999; 341:99-109
14. Anderson U; Wonke B; Percott E; Holden S; Walker JM;Pennell DJ. Comparaison of effects of oral deferiprone and subcutaneous deferrioxamine on myocardial iron concentration and ventricular function in beta-thalassemia. Lancet 2002; 360:516-20 [ Links ]
15. Galanello R; Piga A; Alberti D; Rovan MC; Bigler HP; Secharid R. Safety, Tolerability, and pharmacokinetics of ICL C70, a new orally active iron-chelating agent in patento with transfusion-dependent iron overload due to -thalassemia. JClin Pharmacoll 2003; 43:565-72. [ Links ]
16. Bradai M; Abad MT; Pissand S; Lamraomi F; Skopinski L; Montalemberg M. hydroxyurea can eleiminate transfrusion requeriment in chidren with severe -thalassemia. Blod 2003; 102:1529-30.
17. Boulad F; Giardina P; Gillio A; Keman N; Small T; Brochstein J; Van Syckle K; Szabolcs P; Oreally NJ. Bone marrow transplantation for homozygous beta thalassemia. The memorial sloan-kettering cancer center experience. Ann N. Y. Acad, Sci. 1998; 850: 498-502. [ Links ]
18. May C; Rivella S; Callegari F. Therapeutic haemoglobin synthesis in beta-thalassemia mice expressing lentivirus -enconded human betaglobin. Nature 2000; 406:82-6. [ Links ]
19. Havawa H; Hargrove PW; Kepes S; Srivasta DK; Nienhuis AN; Pearson DA. Extended beta-globin locus control region elements promote consistent therapeutic expression of gamma-globin lentiviral vector in murine beta-thalassemia. Blood 2004; 26: 613-9. [ Links ]
20. Yang B; Kirby S; Lewis J;Detloff PJ; Maeda N; Smithies O. A mouse model -thalassemia. Proc of the nat. Acad Acie. 1995; 92: 11608-12.
21. Pawlik R; Westernnan K; Fabry ME. Correction of sickle cell disease in transgenic mouse models bi gene therapies. Science 2001; 24: 1257-2371. [ Links ]
22. Sáenz GF;Monge B; Arroyo G; Alvarado MA. Enfermedad de Cooley (+ talasemia mayor) en Costa Rica. Sangre, 1976; 1: 117- 122.
23. Rodríguez WE; Jiménez G; Calderón E; Sáenz GF.Beta talasemia mayor (Enfermedad de Cooley) por homocigosidad de gene supresor (º). Rev Cost Cienc Med. 1989; 2: 75-79.
24. Sáenz GF;Chaves M; Grant S; Barrenechea M; Arroyo G; Valenciano E; et al. Hemoglobinas anormales, alfa talasemia y deficiencia de la G6PDEritrocitica en recién nacidos de raza negra. Sangre, 1984; 29: 861-865. [ Links ]
25. Sáenz GF;Jiménez E; Mora L. Enfermedad por Hb H en Costa Rica. Sangre, 1979; 24:277-285. [ Links ]
26. Sáenz GF;Castillo M; Solorzano L; Sánchez G.Talasemia Intermedia. A propósito de un caso de alfa talasemia tipo enfermedad por Hb H. Acta Med Cost., 1984; 27: 179-183. [ Links ]
31. Sáenz GF;Alvarado MA; Atmetla F; Arroyo G;Jiménez J; Valenciano E. "Investigación de hemoglobinas anormales en población costarricense del Guanacaste". Acta Med Cost., 1973; 16:147-153. [ Links ]
32. Hansen EM. Failure to suspect and diagnose thalassemic siyndromes. Arch Intern Med., 11985 45: 93-94. [ Links ]
Ex director del CIHATA-UCR
Abreviaturas: Hb Fetal (Hb F); Hb A2; Hb de Bart (4); genes, cadena de globina; º, +; alelos de talasemia; enfermedad de cooley: genes, cadena de globina; + (l) y º (2) determinantes alélicos de talasemia; Kb kilobase; VCM volumen corpuscular medio-fl; HCM hemoglobina corpuscular media-pg.
 talasemia en todo el Mediterráneo, el Medio Este de Asia, el sudeste Asiático, el subcontinente Indio y otras regiones, indica que sus orígenes genéticos fueron independientes. De igual manera acontece con las alfa talasemias (sudeste asiático, el sur de China, las Filipinas, África y el Mediterráneo). En Costa Rica la alfa talasemia se encuentra en raza negra, con los dos alelos (23% para el
talasemia en todo el Mediterráneo, el Medio Este de Asia, el sudeste Asiático, el subcontinente Indio y otras regiones, indica que sus orígenes genéticos fueron independientes. De igual manera acontece con las alfa talasemias (sudeste asiático, el sur de China, las Filipinas, África y el Mediterráneo). En Costa Rica la alfa talasemia se encuentra en raza negra, con los dos alelos (23% para el  +:(
+:(