Descriptores: genética humana, diagnóstico molecular, FRAXA, cromosoma X, retardo mental hereditario, FMRI, mutaciones inestables.
Abreviaturas: FRAXA: síndrome del cromosoma frágil, CGG: citosina guanina guanina, CpG: dinucleótidos de citidina fosfato guanosina, AGG: adenina guanina guanina, PCR: reacción en cadena de la polimerasa.
Recibido: 18 de diciembre, 2002 ]]>
Aceptado para publicación: 22 de enero, 2002El síndrome de cromosoma X frágil (FRAXA) es la segunda causa genética de retardo mental y la forma más frecuente de retardo mental hereditario, con una frecuencia estimada de 1:4.000 en varones y 1:6.000 mujeres 1. FRAXA es una anomalía hereditaria ligada al cromosoma X, causante de discapacidades que van desde grados variables de problemas de aprendizaje hasta retardo mental. Con frecuencia se asocian retrasos severos en el lenguaje, problemas de conducta, comportamiento semejante al autista, testículos agrandados, orejas grandes o prominentes, hiperactividad, retraso en el desarrollo motor y deficiente integración sensorial 2. Este síndrome es responsable de la mayoría de los casos de trastomos hereditarios en el desarrollo psicomotor. El defecto molecular consiste en una cantidad aumentada de repeticiones de la tripleta citosina guanina guanina (CGG) en una región no codificante del gen FMR1, situado en la porción terminal del brazo largo del cromosoma X. Este síndrome segrega como dominante ligado al X con penetrancia reducida, ya que ambos sexos, cuando acarrean la mutación X frágil, pueden presentar deficiencia mental 3.
En el ámbito molecular, la mayoría de los individuos con este síndrome presentan dos tipos de alteraciones asociadas con el locus FMR- 1:
1. Una de estas alteraciones consiste en la metilación anormal de una región en Xq27.3 rica en dinucleótidos de citidina fosfato guanosina (CpG) región que pertenece a las secuencias reguladores llamadas "islas CpG". La metilación de tales regiones generalmente bloquea la expresión de los genes adyacentes 3. La confirmación de que la metilación anormal se asocia con la manifestación de FRAXA fue la observación hecha por Steinbach y colaboradores 4 de un varón mentalmente normal, portador de una mutación completa pero ausencia de metilación. Estudios posteriores de series de pacientes han mostrado que un 10 a 12% de los varones con un locus FMRI anormal pueden presentar mosaicismo en la metilación, y poseer coeficientes intelectuales normales o casi normales 5. También se ha informado lo contrario, es decir un varón con retardo mental severo, autismo e hiperactividad que al examen molecular mostró tener un alelo normal, de 28 repeticiones pero mosaico para la metilación. La hibridación de Southern presentó en este caso la banda normal para los varones de 2,8 Kb y una banda adicional de 5,2 Kb correspondiente al ADN resistente a la digestión con Eagl, es decir metilado 6.
2. La otra anormalidad encontrada involucro una amplificación de la tripleta CGG en la región 5 del locus FMR-1 7. El análisis del tamaño de esta amplificación en personas normales ha mostrado un ámbito de variación de 6 a 50 repeticiones. En familias afectadas se han encontrado dos tipos de mutaciones 8: las llamadas premutaciones, que no tienen efectos fenotípicos en los portadores, independientemente de su sexo, las cuales involucran amplificaciones entre 52 hasta 200 repeticiones y las mutaciones completas, que se encuentran en los individuos afectados por el síndrome, formadas por más de 200 repeticiones 9.
Esta amplificación es inestable, lo cual se traduce en variabilidad tisular, esto significa que el número de repeticiones es diferente en las células de un mismo individuo; y su tamaño varía también de una generación a otra. La transición del estado de premutación a mutación completa ocurre con una alta frecuencia cuando se trata de la transmisión de una madre portadora a su hijo afectado. La frecuencia con que ocurre esta transición depende del tamaño de la premutación 9,10, y de las tripletas adenina guanina guanina (AGG) intercaladas entre las CGG. Los alelos normales poseen dos tripletas AGG intercaladas, un tercio de los alelos con premutaciones poseen solo una tripleta AGG, y el resto no tienen ninguna. Alelos con repeticiones intercaladas por AGG han mostrado más estabilidad en las transmisiones que los fragmentos constituidos únicamente por CGGs 11.
Excepcionalmente se han encontrado pacientes afectados por FRAXA, que poseen otro tipo de defecto molecular, las mutaciones de punto en las posiciones 304 y 367, cambian residuos del aminoácido isoleucina por asparragina en el dominio KH de la proteína FMRP, dominio importante en la función de la FMRP 3. También se han descrito diferentes tipos de deleciones 12.
La proteína FMRP, producto del gen FMR- 1, está ausente en los pacientes afectados. Su función es participar en el transporte de ARN mensajeros desde el núcleo hacia los poliribosomas principalmente localizados en las dendritas proximales de las neuronas, donde participa en la traducción de las proteínas. Su afinidad es específica para ARNs que se expresan en células nerviosas, incluyendo su propio ARN. El retardo mental es el resultado de anormalidades en la traducción de proteínas que dependen de FMRP para el transporte de sus ARNs y para su traducción 13.
]]> El diagnóstico del cromosoma X frágil se realizaba inicialmente gracias a la expresión citogenética de un sitio frágil en Xq27.3 14 . Esta prueba diagnóstico ha sido relativamente confiable en los laboratorios más eficientes y es efectiva en identificar varones afectados.. En mujeres, el estudio de los cromosomas ha sido mucho más problemático, y la expresión citogenética no es un buen indicador de condición clínica ni es la prueba ideal para portadores 15. Un aumento anormal en el largo de una región de ADN del cromosoma X produce debilitamiento de la cromatina en esa zona, lo cual citogenéticamente se traduce como un sitio frágil. Este sitio frágil se expresa como una laguna isocromatídica en el cromosoma metafásico que se ubica en la posición Xq27.3 7,16. Esta manifestación citogenética, también conocida como marcador, se produce cuando la amplificación de la secuencia del ADN alcanza cierto tamaño.Actualmente existen diferentes sondas específicas para hacer diagnóstico directo de la mutación en los afectados mediante la hibridación de Southern. Con esta técnica es posible determinar amplificaciones de 80 o más repeticiones. Cuando los familiares de un individuo afectado demandan consejo genético, se hace necesario usar la reacción en cadena de la polimerasa (PCR), para diagnosticar los portadores de amplificaciones pequeñas, lo cual permite conocer exactamente el tamaño de la región amplificada. Combinando los dos métodos es posible discriminar entre el alelo normal, el alelo premutado y el alelo con la mutación completa.
Las alteraciones fenotípicas que produce esta amplificación del ADN son diversas, y se enmarcan dentro de un síndrome bien caracterizado llamado síndrome de cromosoma X frágil o síndrome de Martin-Bell, en honor a J.P. Martin y a J. Bell, quienes delinearon el síndrome por primera vez en 1943 (una detallada descripción se encuentra en Castro & Cuenca, 1996).
FRAXA ha sido detectado en todos los grupos étnicos mayoritarios 3. El estimado de prevalencia total del síndrome, obtenido de estudios moleculares en Inglaterra varía de 1: 2 700 a 1: 5 700 varones 17 y es de 1: 4 350 en Australia 1. El análisis molecular ha permitido ahora el estudio de la prevalencia de premutaciones en mujeres. Un estudio sistemático realizado en mujeres de la población general de Quebec encontró que 1:500 es portadora de un alelo mayor a las 66 repeticiones, y que 1:500 es portadora de un alelo con un tamaño entre 55 y 63 (zona gris). Estas últimas tienen un riesgo menor de sufrir inestabilidad meiótica que las primeras, por lo que tienen un riesgo menor de tener un niño afectado 18.
Mientras no se pueda "curar" la condición de portador de la mutación, la prevención primaria de la ocurrencia de esta patología se basa en la asesoría genética adecuada y oportuna a los miembros de la familia afectada. Como ya hemos explicado, las limitaciones en este sentido que impone el diagnóstico únicamente citogenético, se logran superar gracias al análisis directo de la mutación mediante técnicas de amplificación e hibridación del ADN. Un estudio en Nueva Gales del Sur ha demostrado que en diez años de consejo genético se ha logrado bajar la incidencia de FRAXA a la mitad y restaurar la confianza reproductiva a los miembros de las familias afectadas 20 . La prevención secundaria o post-concepción se logra en los países donde se permite la interrupción del embarazo, mediante diagnóstico precoz, embrionario o fetal. Aplicando las técnicas moleculares mencionadas, se ha conseguido el diagnóstico molecular, más exacto que el que se obtenía mediante el uso de técnicas citogenéticas 21. En cuanto a prevención terciaria, el estudio a fondo de los pacientes con éste síndrome ha permitido plantear estrategias específicas para mejorar su funcionamiento intelectual y su adaptación social 22, de ahí la importancia de la confirmación molecular del diagnóstico clínico.
El tratamiento es sintomático mientras no se cuente con terapia de reemplazo de la proteína o terapia génica de este síndrome 23,24.
El enfoque más útil en el tratamiento del síndrome X frágil es el multidisciplinario y de conjunto, que debe incluir un maestro de educación especial, un terapeuta de lenguaje, un terapeuta ocupacional, un médico, un psicólogo y un consejero genético 2.
Con el fin de contribuir a que los pacientes costarricenses afectados por FRAXA y sus familias puedan beneficiarse de las metodologías modernas que ofrece la biología molecular este trabajo tuvo como objetivos:
Establecer la presencia de mutaciones inestables en el gen FMRI, determinar el estado de metilación del mismo y la cantidad de repeticiones de la tripleta CGG en las personas portadores de premutación.
Asesorar a las familias en las que segrega esta patología para que conozcan lo mejor posible esta enfermedad (causa, consecuencias, pronóstico, posibilidades de tratamiento y/o rehabilitación, manejo apropiado de las secuelas concomitantes, riesgos de ocurrencia o de recurrencia, etc.), ayudarlas a adaptarse positivamente y estimular el abordaje preventivo del síndrome y de sus secuelas.
]]> Fomentar el conocimiento de esta patología entre el personal de salud y entre las personas encargadas de la educación de estos niños, para contribuir a potencializar sus fortalezas, desarrollar sus áreas débiles y manejar adecuadamente sus problemas de conducta y de adaptación social. Este trabajo muestra los primeros esfuerzos en Costa Rica para hacer diagnóstico directo de la mutación en niños bajo sospecha clínica de FRAXA y sus familiares cercanos.
Materiales y métodos
El protocolo de esta investigación fue conocido y estudiado por el Comité de ética del Hospital Nacional de Niños. Los pacientes estudiados se clasificaron en: grupo uno, 13 probandos con examen citogenético positivo, ya sea estudiados previamente en el INISA o en otros laboratorios; grupo dos, N = 30, sus familiares cercanos y el grupo tres, constituido por niños referidos por maestros, pediatras y sicólogos ( N = 15). Siempre que un niño o niña fué referido para estudio, se procedió a explicar a los padres las características del síndrome y los beneficios que la familia podía recibir a través del estudio molecular. Se obtuvo el consentimiento informado, se construyó la genealogía de la familia y se invitó a participar a otros familiares.
Se obtuvo una muestra de sangre en anticoagulante ACD, se extrajo el ADN según el método tradicional 25, se cuantificó y se realizaron digestiones con la, o las enzimas de restricción respectivas: Hind III o Eco RI y Eagl, dependiendo del tipo de hibridación a realizarse. En cada individuo se llevó a cabo una hibridación utilizando la sonda Ox. 1.9 26 o la StB 12.3 27 posterior a la digestión sencilla con Hind III, para determinar el tamaño de la amplificación. Simultáneamente se llevó a cabo una hibridación con la sonda StB 12.3 posterior a una doble digestión, en la cual, además de determinar el tamaño, por adición de la enzima Eagl que discrimina el estado de la metilación, se determinó si el gen está activo o inactivo.
Con el fin de poder ofrecer la mejor información a los potenciales portadores de premutaciones se utilizó la reacción en cadena de la polimerasa (PCR), y la posterior electroforesis de los productos en geles de poliacrilamida desnaturalizantes, según el método descrito por Fu 9 en 35 familares de pacientes. También se hizo control de calidad de los resultados de la PCR mediante el uso de un analizador automático de ADN en 19 pacientes 29.
Una vez obtenidos los resultados moleculares se hicieron sesiones de consejo genético, en las cuales se explicó detalladamente y en lenguaje sencillo los resultados y las consecuencias en cada persona estudiada.
Resultados
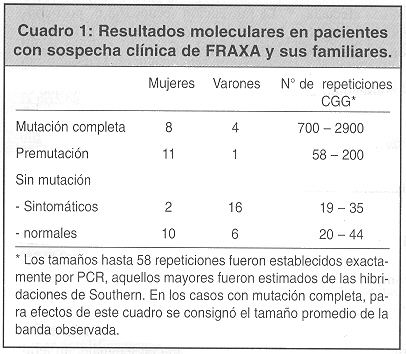
]]> Utilizando la metodología descrita, los individuos normales presentan una banda de 2,8 Kb cuando se utiliza la enzima Hind III y la sonda Ox1.9, bandas de mayor tamaño corresponden a amplificaciones. Cuando se usa la doble digestión y la sonda StB 12.3; según se puede observar en la figura 1; los varones normales presentan una banda de 2,8 y las mujeres normales presentan dos bandas, la de 2,8 correspondiente al alelo localizado en el cromosoma X activo y la de 5,2 correspondiente al alelo localizado en el cromosoma X inactivo y metilado (por lo tanto resistente a la digestión con la enzima Eag l). Las mujeres portadoras de premutaciones pequeñas pueden tener bandas de mayor tamaño en cualquiera de los dos alelos. Sin embargo las portadoras de mutaciones completas generalmente presentan aumentado de tamaño el alelo de 5,6 ya que las amplificaciones grandes promueven la metilación.De esta manerra, se obtuvieron 58 estudios moleculares de niños con sospecha clínica o con diagnóstico citogenético del síndrome del cromosoma X frágil y de familiares cercanos de los niños con resultados positivos (cuadro 1).
En nueve niños del grupo uno se confirmó el diagnóstico de mutación completa del gen FMR1, en los otros cuatro se descartó al encontrarse resultados normales en todas las pruebas moleculares. Los nueve niños positivos habían sido diagnosticados mediante estudio de cromosomas en el INISA, los niños negativos son cuatro hermanos diagnosticados en otro laboratorio fuera de la U.C.R. Los niños negativos para la mutación FRAXA se estudiaron también para FRAXE, otro retardo mental asociado a un marcador de fragilidad en el cromosoma X, resultando también negativos. Entre los familiares de estos niños (grupo 2) se encontraron dos mujeres con la mutación completa y doce personas con la premutación: un varón transmisor normal y once mujeres portadoras. El resto de los familiares (N=16) obtuvo resultados normales. En el grupo tres se detectó una niña con la mutación, el resto fueron normales, tanto mediante estudios citogenéticos como moleculares.
En la figura 2 se muestra la genealogía de una de las familias estudiadas, donde se puede observar que la mutación se inició en el abuelo, quien porta una premutación pequeña, de 58repeticiones, pero con capacidad de amplificarse, dando como resultado tamaños mayores en las hijas (II-1; II-3 y II-9 con amplificaciones entre las 65 y 70 repeticiones; II-12 con 160 repeticiones y la II-8 con 62 repeticiones), en quienes el alelo siguió creciendo, posibilitando de esta manera el nacimiento de varoncitos afectados con retardo mental.
Este trabajo representa el primer informe en Costa Rica sobre resultados de estudios moleculares en pacientes afectados por el síndrome del cromosoma X frágil y sus familiares. Estos estudios, han pasado a formar parte de la rutina en el diagnóstico y manejo de estos pacientes y sus familiares, desde comienzos de la década de los noventa, cuando se obtuvieron las sondas de ADN específicas para el gen FMRI, en los países desarrollados y algunos países en vías de desarrollo.
]]> Los resultados de este estudio demuestran la importancia de confirmar la sospecha clínica y/citogenética con la biología molecular antes de ofrecer el consejo genético a los pacientes ya que encontramos un diagnóstico negativo para la amplificación de] gen FMRI en cuatro niños que fueron referidos por presentar el marcador cromosómico. El uso de la e¡togenética es limitado a los afectados por mutaciones completas y tiene el riesgo de arrojar resultados falsos positivos, que confundirán tanto al consejero como a la familia. El hallazgo de resultados moleculares negativos para FRAXA en niños con retardo mental de causa desconocida y positivos para el marcador cromosómico es común en la literatura debido a la presencia de otros sitios frágiles en la porción distal del brazo largo del eromosoma X 29.La prevención basada en el consejo genético es la forma más efectiva de enfrentar este síndrome. En Nueva Gales del Sur, Australia, (con una población de 6,5 millones de habitantes) por ejemplo, la tasa de incidencia ha bajado diez veces, de 4,3/10.000 a 0,5/10.000 como consecuencia del programa de consejo genético, apoyado en diagnóstico prenatal e interrupción del embarazo 20.
En Costa Rica, no existen datos sobre la inversión económica y social que representa para la sociedad y las familias afectadas, la atención de las personas discapacitadas. Sin embargo los pacientes afectados por FRAXA requieren atención y dedicación de otras personas durante toda su vida.
La esperanza de vida de los afectados y portadores es igual que la población general, las mujeres portadoras de premutación presentan un riesgo mayor de padecer menopausia precoz y trastornos efectivos.
Los niños oportuna y correctamente diagnosticados con el síndrome del cromosoma X frágil se benefician con la adquisición de este conocimiento por parte de sus padres y de sus maestros ya que tienen características particulares que los diferencian de otros niños con retardo o dificultades de aprendizaje de otro origen. Estas peculiaridades hacen necesario un abordaje psicopedagógico especial, el cual ha tenido éxito probado en otros países y ha contribuido al desarrollo de los niños de manera que funcionen con la mayor independencia posible, en ambientes tan normales como es factible 29. Por otro lado, la identificación de personas portadoras en las familias les permite tomar decisiones en cuanto a reproducción acorde a sus necesidades, circunstancias y valores. Además para las personas sin esta mutación, el alivio de su preocupación respecto al riesgo de transmisión es grande.
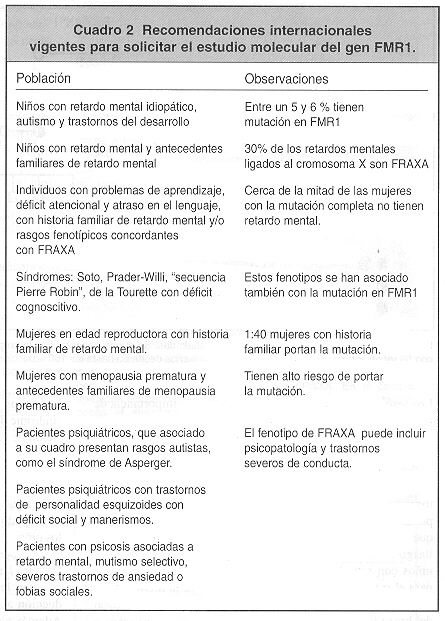
En el cuadro 2 se resumen las recomendaciones internacionales vigentes para referir a los pacientes al laboratorio para estudio molecular del gen FMRI.
El Instituto de Investigaciones en Salud ofrece a la comunidad médica nacional y a las escuelas de enseñanza especial el servicio de diagnóstico molecular, y en los casos que ameriten las sesiones de consejo genético que sean necesarias con los familiares.
Agradecimientos
Al Prof.Dr. Jean-Luis Mandel de la Universidad Luis Pasteur de Estrasburgo, Francia, por permitimos el uso de la sonda StB 12.3 y al Prof. Dr. Key Davies del Instituto de Medicina Molecular de Oxford, Inglaterra, por permitirnos usar su sonda Oxl.9. Al Organismo Internacional de Energía Atómica , la Comisión de Energía Atómica de Costa Rica y a la Vicerrectoría de Investigación de la Universidad de Costa Rica, proyecto N° 742-95-257, por su apoyo financiero. A la señora Regina Neumann y al señor Federico Hernández por su apoyo técnico. A los Dres. Peter Nümberg, Hartmut Peters, Peter Steinbach y Douglas Wilcox por su colaboración.
Abstract
Fragile X syndrome is the most common hereditary type of mental retardation, affecting 1:4 000 males and 1:6 000 females. Unfortunately, most persons with this syndrome have not bcen diagnosed and are classified as cases of mental retardation of unknown origin. The correct etiological classification of these patients would allow their families lo avoid recurrence of this disease through adequate genetic counseling. As a result, this study intended to provide accurate molecular diagnosis of fragile X syndrome in mentally retarded patients with clinical or cytogenetic suspicion of the disease, to detect the normal transmitting males and female carriers in each of the proband's families and to promote prevention after proper genetic counseling. To achieve this, genomic DNA was digested with Hind 111, EcoRI and Eagl and Southern blotting was performed with probes Oxl.9 and StB 12.3. To asses the size of the trinucleotide repeat, PCR was used in some of the cases. Three groups were studied: group one with 13 children with the cytogenetic marker, 30 of their close relativas conformes group two and group three with 15 clinically suspicious fragile X syndrome children. Results: of the 13 probands, four proved not to be fragile X cases since their average repeat number was 30. In group two, two females with the full mutation, one normal transmitting male and eleven female carriers with the premutation were found. In group three an additional female with the full mutation was identified, the rest had normal DNA and cytogenetic test results. In summary, DNA studies are a better way to accurately asses the full mutation and premutation carriers. The right diagnosis renders benefits to fragile X cases in terms of the interventions they need and to carriers since this information is a must for proper genetic counseling and prevention. Moreover, molecular studies are cheaper than cytogenetie diagnosis in well equipped laboratories with trained personnel.
]]>
1. Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am.J.Med Genet. 1996: 64:196-97. [ Links ]
2. Hagerman R. Clinical and diagnostic aspects of fragile X syndrome.
En: Wells R Warren S T, Sarmiento M, ed. Genetic instabilities and hereditary neurological diseases. New York: Academic Press. 1998: 15-25.
3. Imbert G, Feng Y, Nelson D L, Warren S T, Mandel. 1998. FMRI and mutations in fragile X syndrome: Molecular biology, biochemistry, and genetics. En: Wells R Warren S T, Sarmiento M ed. Genetic instabilities and hereditary neurological discases. New York: Academie Press. 1998: 27-54. [ Links ]
4. Wührle D, Salat U, Gláser D, Mücke J, Meisel-Stosiek M, Schindler D, et al. Unusual patterns of mutations in high functioning fragile X males and other cases may result from instability of expanded CGG repeats in the absence of methylation. J.Med.Genet. 1998: 35:103-111. [ Links ]
5. Lachiewicz A M, Spiridigliozzi G A, MeConkie-Rossell A, Burhess D, Feng Y, Warren S T et al. A fragile X male with a broad Smear on Southern blot analysis representing 100-500 CGG repeats and no-methylation at the Eagl site of the FMR- 1 gene. Am. J. Hum. GMet. 1996: 64: 278-282. [ Links ]
6. Battagia P, de Fanti, E, Grimau-Merino R, Giardina L, Schiavon R. Laboratory findings in a male with suspected X-fragile syndrome; Discovery of mosaicism of normal and methylated FMR-1 genes. Eur. J. Hum. Genet. 2001: 9, Supl 1: 172 [ Links ]
7. Verkerk A J M, Pieretti M, Sutcliffe J S, Fu Y H, Kuhl D P, Pizzuti A, et al. Identification of a gene (FMR-1) contain ing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell: 1991:65 :905-914. [ Links ]
8. Oberlé I, Rosseau E, Heitz D, Kretz C, Deveys D et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. 1991:Science. 252:1097-1102. [ Links ]
9. Fu Y H, Kuhl D P A, Pizutti A, Pieretti M, Sutcliffe J S, Richards S et al. Variation of the CGG repeat al the fragile X site results in genetic , instability: resolution of the Sherman paradox. Cell. 1991: 67 :1047-1058 [ Links ]
10. Yu S, Pritchard M, Kremer E, Lynch M, Nancarraw J, Baker E, et al. Fragile X genotype characterized by an unstable region of DNA. Science. 1991: 252:1179-81. [ Links ]
11. Eichler E E, Holden J J A, Popovich B W, Reiss A L, Snow K, Thibodeau S N, et al. Length of uninterrupted CGG repeats determines stability in the FMR1 gene. Nature Genet. 1994: 8: 88-94 [ Links ]
12. Quan E, Zonana J, Gunter K, Peterson K L, Ma,,enis R.E. Popovich B W. An atipical case of fragile X syndrome calised by a deletion that includes the FMRI gene. 1995: Am.J.Hum.Genet. 56 :1042-1051. [ Links ]
13. Jin P, Warren ST. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet. 2000: 9:901-908. [ Links ]
14. Castro l, Cuenca R. Frecuencia del síndrome del cromosoma X frágil en la Escuela de Enseñanza Especial "Fernando Centeno Güell" Act Ped Cost 1996:10(2):99-106. [ Links ]
15. Tommerup N. Cytogenetics of the fragile site at Xq27. En: Davies K E. ed. The fragile X syndrome. Oxford: Oxford University Press. 1989:102-135. [ Links ]
16. Krawcsun M S, Jenkins E C, Brown W T. Analysis of the fragile X chromosome: localization and detection of the fragile site in high resolution preparation. Hum Genet 1985: 69:209-21 1. [ Links ]
17. Morton JE, Bundey S, Webb TP MacDonald F, RindI PM, Bullock S. Fragile X syndrome is less common than previously estimated. J Med Genet. 1997:34:1-5 [ Links ]
18. Rousseau F, Rouillard P, Morel M L, Khandjian E W, Morgan K. Prevalence of carriers of premutation.size alleles of the FMRI gene and implicaúons for the population genetics of the fragile X syndrome. Am J Hum Genet 1995:57: 1006-1018.
]]>19. Falik-Zaccai T C, Shachak E, Yalon M, Lis Z, Borochowitz Z, MacPherson J N , et al. Predisposition to the fragile X syndrome in Jews of Tunisian descent is due to the absence of AGG interruptions on a rare Mediterranean haplotype. Am J Hum Genet 1997:60:103-112. [ Links ]
20. Turner G, Robinson H, Wake S, Laing S, Partington M. Case finding for the fragile syndrome and its consequences. B M J 1997:315: 1223-1226. [ Links ]
21. Sutherland GR, Gedeon A, Korman L, Donnelly A, Byard RW, Mulley J C et al. Prenatal diagnosis of fragile X syndrome by direct detection of unstable DNA sequence. N Engl J Med 1991: 325: 1720-1722. [ Links ]
22. Spiridigliozzi G A, Lachiewicz M, MacMurdo C S, Vizoso A D, O'Donnel C M, McConkie-Rossel A, et al. Educating boys with fragile X syndrome. A Guide for parents and professionals. Duke University Medical Center. 1994: 1-20 [ Links ]
23. Hagerman R J. Medical follow-up and pharmacotherapy. En: Hagerman R J , Cronister A eds. Fragile X syndrome diagnosis, treatment and rescarch. Baltimore: The John Hopkins University Press. 1996. 3-250. [ Links ]
24. Sobesky W E. The treatment of emotional and behavioral problems.
En: Hagerman R J , Cronister A cds. Fragile X syndrome diagnosis, treatment and research. Baltimore. The John Hopkins University Press. 1996: 332-340.
25. Strauss W M. Preparation of genomic DNA from mammalian tissue. En: Ausubel F M, Brent R, Kinston R E eds. Current protocols in molecular biology. New York: John Wiley and Sons Inc. 1988:1:2.2.1-2.2.3 [ Links ]
26. Snow K, Doud L, Hagerman R, Hull C, Hirst M C, Davies K E, et al. Analysis of mutations at the fragile X locus using the DNA probe Ox1.9. Am J Med Genet 1992:43:244-254. [ Links ]
27. Rousseau F D, Heitz D, Biancalana S, Blumenfeld C, Kretz J, Boué J, et al. Direct diagnosis by DNA Analysis of the fragile X syndrome of mental retardation. N Engl J Med 1991:325( ):1673-1681. [ Links ]
28. Larsen L A, Gronskov K, Norgaard-Pedersen B, Brondum-Nielsen K, Hasholt L, Vuust J. High-throughput analysis of fragile X (CCG)N alleles in the normal and premutation range by PCR amplification and automated capillary electrophoresis. Hum Genet 1997:100: 564-568 [ Links ]
29. Arrieta I, Criado B, Martínez B, Telez M, Nuñez T, Panagarikano O, et al. A survey of fragile X syndrome in a sample from Spanish Basque country. Ann Genet 1999:42:197-201. [ Links ]
30. Braden M. Fragile, handle with care. Understanding fragile X syndrome. Chapel Hill: Avanta Publishing Company. 1996:1-169. [ Links ]
1 Instituto de Investigaciones en Salud (INISA),
2 Escuela de Biología,
3 Escuela de Medicina, Ciudad Universitaria Rodrigo Facio, San José Costa Rica.
Correspondencia: Dra. Patricia Cuenca, INISA, Universidad de Costa Rica, 2060 San José; fax: (506) 207-5130;
correo electrónico: pcuenca@cariari.ucr.ac.cr