









Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAgronomía Mesoamericana
versão On-line ISSN 2215-3608versão impressa ISSN 1659-1321
Agron. Mesoam vol.24 no.1 San Pedro Jun. 2013
Variabilidad genética de aislamientos colombianos del potato mop-top virus (PMTV)
Genetic variability of colombian isolates of Potato mop-top virus (PMTV)
Genetic variability of colombian isolates of Potato mop-top virus (PMTV)
*Dirección para correspondencia:
Resumen
El objetivo de este trabajo fue evaluar los niveles de variación de aislamientos de PMTV en cultivos de papa de Colombia durante los años 2010 y 2011. Se obtuvieron secuencias de los genes de la cápside viral (CP, ARN 2) y del Triple Bloque de Genes (TGB2, ARN 3) de cepas de PMTV de los departamentos de Antioquia, Boyacá, Cundinamarca y Nariño. Adicionalmente, dos de los aislamientos fueron secuenciados en >83% de sus ARN 2 y 3. Los análisis filogenéticos basados en CP indicaron la presencia de dos clados. El primero contiene cepas de referencia mundial de PMTV en conjunto con 19 de las cepas de este estudio; mientras que el segundo clado sólo agrupó cepas de Colombia, las que compartieron menos de 76% de identidad con el primer grupo. Esto posiblemente indica la ocurrencia de una nueva especie de pomovirus, aunque se requiere secuenciación de todo el genoma para confirmar dicha hipótesis taxonómica. El análisis del TGB2 generó un solo clado, agrupando indistintamente las cepas colombianas de PMTV con las de otros países. El análisis de los segmentos 2 y 3 del genoma viral, indicó que los dos aislamientos colombianos secuenciados compartían >94% de identidad con las secuencias de cepas de República Checa y Suecia. Estos resultados pueden ser utilizados para el diseño de herramientas de diagnóstico del PMTV en los Andes, que apoyen los programas de certificación de tubérculo-semilla y mejoramiento genético de papa.
Palabras clave: RT-PCR, secuenciación, Solanum tuberosum.
Abstract
The objective of this work was to study the level of variation of PMTV isolates from Colombian potato crops through 2010 and 2011. Genes encoding for the viral coat protein (CP, ARN 2) and the Triple Gene Block (TGB2, ARN 3) were sequenced from PMTV isolates obtained in the provinces of Antioquia, Boyacá, Cundinamarca y Nariño. Additionally, RNA 2 and RNA 3 from two isolates were almost sequenced to completion (>83%). Phylogenetic analysis based on the CP sequence revealed the presences of two clades. The first clade included PMTV reference strains from all over the world and 19 Colombian isolates used in this study. The second class grouped only Colombian isolates and shared less than 76% identity with clade 1, suggesting a new pomovirus species. Complete genome sequencing is required to confirm this hypothesis. Phylogenetic analysis using the TGB2 gene grouped all sequences in a single clade. Sequence analysis of segments 2 and 3 of the viral genomes revealed >94% sequence identity with PMTV isolates from Czech Republic and Sweden. These findings will be helpful in the development of diagnostic tools for PMTV in the Andes in order to support tuber-seed certification and genetic improvement programs.
Key words: RT-PCR, sequencing, Solanum tuberosum.
Introducción
El Potato mop-top virus (PmTV), es la especie tipo del género Pomovirus y recientemente se ha clasificado en la nueva familia Virgaviridae (adams et al. 2009). Su ámbito de hospedantes incluye un número limitado de especies de las familias solanaceae y Chenopodiaceae, siendo la papa (Solanum tuberosum L.) su principal hospedante desde el punto de vista económico (Kirk 2008). PMTV es transmitido por tubérculo semilla infectado y por zoosporas del plasmodiofórido Spongospora subterranea f. sp. subterranea (Wallr.) (Lagerh) (Sss), agente causal de la sarna polvosa de la papa (Harrison y Jones 1970). Su presencia se ha registrado principalmente en regiones con climas húmedos y fríos como el Norte de Europa, las islas Británicas, Estados Unidos y Canadá, Japón, las zonas montañosas de Costa Rica y los Andes (Xu et al. 2004, salazar 2006, montero-astúa et al. 2008, Nakayama et al. 2010, santala et al. 2010). a pesar de que se considera que PMTV es originario de los andes, su ocurrencia en colombia solo fue detectada en el año 2007 (Vélez 2007) y reconfirmada por Gil et al. (2011).
El PMTV puede causar pérdidas de hasta 26% en el rendimiento del cultivo de la papa, lo que sumado al efecto del daño ocasionado por su vector Sss, pudiera ascender a niveles de 50 a 80% (Jones y Harrison 1972, Guerrero 2000). Sin embargo, dichos niveles de pérdidas dependen de la susceptibilidad de las variedades cultivadas (Xu et al. 2004, santala et al. 2010), las variantes del patógeno (Harrison y Jones 1970, nielsen y nicolaisen 2003) y fundamentalmente de las condiciones ambientales prevalentes durante su infección (Davey 2009, Latvala-Kilby et al. 2009).
El PMTV es un virus tripartita con morfología de varilla rígida de 18-20 nm en diámetro y longitudes de 290-310 nm, 150-160 nm y 65-80 nm. cada partícula contiene una molécula de ARN de cadena sencilla con polaridad positiva (ARNss+). El ARN 1, de 6043 nucleótidos (nt) codifica para una proteína que tiene motivos de metiltransferasa y helicasa (ORF 1) y una proteína generada por supresión del codón de finalización del ORF 1 con motivos de ARN polimerasa ARN dependiente (RdRp) (Savenkov et al. 1999, 2003). el ARN 2 (3134 nt) contiene un ORF para la proteína de la cápside (CP) de 20 kDa. También codifica para una proteína de 90 kDa generada por lectura continua (readthrough, CP-RT), a través de un codón ámbar en el gen de la CP. Diversos estudios han indicado que esta proteína RT está involucrada en la transmisión por sss del PmTV, siendo demostrada mediante análisis de inmuno-marcaje, su localización en los extremos de las partículas virales (Cowan et al. 1997).
El ARN 3 (2694 nt) codifica para cuatro polipéptidos de 51, 21, 13 y 8 kDa, respectivamente. Los primeros tres presentan secuencias similares a proteínas del Triple Bloque de Genes (TGB) involucradas en el movimiento de célula a célula de algunos virus de plantas. Las TGBs de los pomovirus corresponden a la clase 1 o grupo hordei de estas proteínas, en conjunto con las codificadas por otros géneros de la familia Virgaviridae y del género Hordeivirus. Se cree que TGB2 y TGB3 asisten a TGB1 en su función de trasportar el genoma viral a través de los plasmodesmos (Lim et al. 2009). La cuarta proteína (8 kDa) es rica en cisteína (CRP) y aparentemente está involucrada en el movimiento sistémico del PMTV y en la expresión de síntomas necróticos en hospedantes experimentales (Savenkov et al. 1999, 2003).
Los síntomas que causa PMTV en papa se manifiestan por agrietamientos en la superficie de los tubérculos, pudiendo estar acompañados por la presencia de anillos necróticos en la corteza e incluso la parte interna del mismo, síntoma conocido como ¨Spraing¨ (Harrison y Jones 1970). En las hojas de las plantas infectadas se puede observar un moteado en forma de V tipo “aucuba”, además de la deformación del follaje y el acortamiento de los entrenudos (mop-top) (Xu et al. 2004). En la región Andina, no es frecuente observar la sintomatología en los tubérculos ni los moteados tipo ¨aucuba¨, no siendo posible asociar síntomas específicos con la presencia de PMTV, situación que explica en buena parte el porqué del desconocimiento de su efecto sobre las variedades sembradas en esta región suramericana (Tenorio et al. 2006, gil et al. 2011). sin embargo, según Salazar (2006), el PMTV es uno de los virus prevalentes en el cultivo de la papa en los Andes.
A nivel mundial, se ha reportado la presencia de bajos niveles de variación entre genotipos de PMTV (mayo et al. 1996, santala et al. 2010). Latvala-Kilby et al. (2009), comparando secuencias de cP de 23 aislamientos de Finlandia y Letonia con respecto a secuencias disponibles en GenBank para cepas de Escocia, Dinamarca, Suecia y República Checa, encontraron identidades superiores al 98% entre estas, con solo siete diferencias a nivel de aminoácidos (aa) entre todas las comparaciones. Las evaluaciones de secuencias de CP-RT, también presentaron altos niveles de identidad (98 a 100%), aunque en este caso 24 posiciones en las secuencias de aa fueron variables. resultados similares reportaron mayo et al. (1996) al comparar las secuencias de cP de tres aislamientos de Escocia y ocho de Perú; mientras que Xu et al. (2004), encontraron que seis de las secuencias de CP de aislamientos de PMTV de Norteamérica presentaron identidades superiores al 97% con respecto a secuencias de aislamientos de diferentes países de europa. a pesar de estos bajos niveles de variación, cuando se realizan análisis filogenéticos con dichas secuencias, se han encontrado dos clados que dividen los aislamientos de PMTV y que pueden ser identificados por análisis de RFLPs (Nielsen y Nicolaisen 2003).
En Colombia, Vélez (2007), encontró por lo menos dos genotipos de PMTV definidos a partir de la secuencias del gen CP: un grupo que presentaba altos niveles de identidad con respecto aislamientos de Europa y Canadá, y otro grupo constituido exclusivamente por aislamientos colombianos. Esta situación fue confirmada posteriormente por Gil et al. (2011), quienes encontraron que las variantes de PMTV tan solo presentaban un 76% y 86% de identidad para cP y TgB2 y con respecto a los aislamientos mundialmente reportados de PMTV. Sin embargo dichos análisis fueron conducidos con un bajo número de secuencias (cuatro para CP y tres para TGB2) debido a las dificultades experimentales que representa el trabajo con este virus (bajo título en plantas de papa, distribución errática, ausencia de cebadores específicos para cepas andinas, etc).
El objetivo de este trabajo fue evaluar los niveles de variación de aislamientos de PMTV en cultivos de papa de Colombia durante los años 2010 y 2011. El propósito fue ampliar el número de secuencias nucleótidas para la cP y TgB2 de aislamientos de PmTV obtenidos en diferentes regiones productoras de papa de Colombia, de manera que fuera posible determinar si la presencia de las variantes de este virus corresponde a una situación generalizada o por el contrario responde al efecto de muestra dado el bajo número de cepas analizadas por Vélez (2007) y gil et al. (2011). Adicionalmente, se realizó la secuenciación de gran parte de los ARN 2 y ARN 3 de dos cepas de PMTV obtenidas en los Departamentos de Antioquia y Boyacá (Colombia), realizándose un análisis de variación con respecto a las secuencias de referencia de genomas completos disponibles en las bases de datos moleculares.
Materiales y Métodos
Colección y preparación de muestras
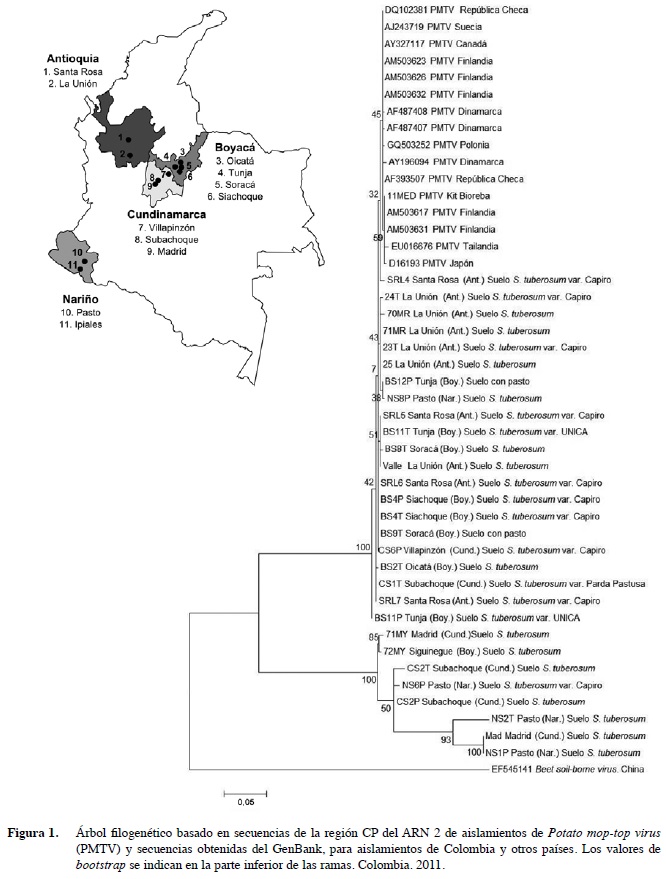
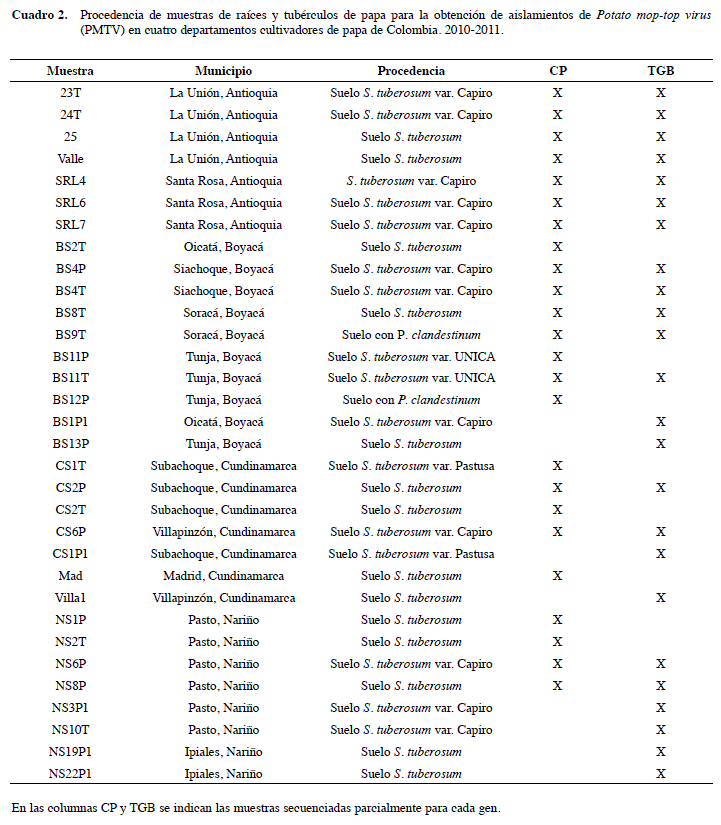
Se obtuvieron muestras de raíces y tubérculos de papa con síntomas de sarna polvosa, así como de suelos de lotes con reportes de la presencia de sss, en las zonas colombianas productoras de papa de los municipios de la Unión y Santa Rosa de Osos (Antioquia); Zipaquirá, Tabio, Subachoque y Villapinzón (Cundinamarca); Tunja, Siachoque, Soracá y Oicatá (Boyacá) y Pasto e Ipiales (Nariño) (Figura 1). Para la separación de los quistosoros del suelo, se siguió la metodología de Jaramillo y Botero (2007), en la cual las muestras de suelo (500g) son pasadas por un juego de tamices de 100 y 25 µm. Aproximadamente, 1 g de los quistosoros obtenidos fueron inoculados en Nicotiana benthamiana y Solanum phureja, utilizadas como plantas señuelo para incrementar el inóculo de PMTV a partir de la infección de su vector Sss (Gil et al. 2011). El material vegetal fue mantenido en macetas con 250 g de turba como sustrato, bajo condiciones de casa de malla en el Centro Experimental Paysandú, de la Universidad Nacional de Colombia (sede Medellín) ubicada en el corregimiento de Santa Elena, municipio de Medellín. La zona de vida es bhMB (Holdridge 1987), con una altitud de 2550 msnm, temperatura media 14°C y precipitación promedio anual de 2000 mm. Tres meses después de la siembra, se recolectaron las hojas y raíces de las plantas señuelo para su procesamiento en el laboratorio.
Secuenciación de CP y TGB2
Se obtuvo el ARN total de plantas de N. benthamiana y S. phureja mediante el RNeasy plant mini kit, a partir de 100 mg de tejido, utilizando 450 μl de buffer RLT para tejido foliar o RLC para raíces y 4,5 μl de β-mercaptoetanol, siguiendo las instrucciones del fabricante. Al finalizar el procedimiento, el ARN obtenido se eluyó en 40 μl de agua destilada estéril tratada con DEPC.
Las reacciones de RT-PCR se realizaron en dos pasos. Para la retrotranscripción (RT) se utilizó un volumen final de 20 µl, incluyendo 0,5 µl de agua destilada estéril, 1,5 µl de PBST (0,5%), 4 µl de buffer RT (5X), 4 µl de MgCl2 (25 mM), 2 µl de dNTPs (10 mM), 2 µl del cebador reverso específico 123_end (10 µM) (Savenkov et al. 1999), 0,5 µl de inhibidor de ARNasas (40U/µl), 0,5 µl de retrotranscriptasa del virus de la leucemia murina (M-MuLV) (20 U/µl) y 5 µl de ARN o alternativamente una dilución 1:5. El programa de RT fue de 37°C por 60 min, seguido de 70°C por 10 min, para inactivar la enzima. Las muestras del ADN copia (ADNc) se almacenaron a 4°C hasta su uso posterior. Como control positivo de amplificación de PMTV, se utilizó la muestra proveída para este fin por el Kit de ELISA de Bioreba, realizándose la extracción de RNA en la misma forma que para las demás muestras.
Para las reacciones de PCR se emplearon los cebadores específicos H360 y C819 (MacKenzie 1996) para amplificar una región de 459 pb del gen CP entre las posiciones 385 y 844 del genoma viral; así como los cebadores PMTVF4 y PMTVR4 para obtener amplicones de 415 pb del TgB2, entre las posiciones 1726 y 2141 del ARN 2 (Xu et al. 2004). Para algunos aislamientos en los que se dificultaba la amplificación del TGB2, se utilizaron los cebadores PMTV5 y PMTV7 (Lambert et al. 2003) que amplifican un fragmento de 646 pb y se extienden entre las posiciones del genoma 1417 a 2063. Las PCR se realizaron en 25 µl incluyendo 15,8 µl de agua destilada estéril, 2,5 µl del amortiguador para la enzima (10X), 1,8 µl de MgCl2 (25 mM), 0,5 µl de dNTPs (10 mM), 0,5 µl de cada cebador (10 µM), 0,2 µl de albúmina de suero bovino (20 mg/ml), 0,2 µl de Taq ADN polimerasa (5 U/µl) y 3 µl de ADNc. El programa consistió de una desnaturalización inicial a 98°C por 3 min, seguida por 40 ciclos de 94°C por 30 s, 53°C por 45 s, 72°C por 1 min y un período final de extensión a 72°C por 10 min. Luego de la amplificación, se tomaron 5 µl de los productos de reacción y se separaron por electroforesis en gel de agarosa al 1,5% suplementado con bromuro de etidio (10 mg/ml) y su tamaño se verificó por comparación con el marcador de peso molecular Generuler 100 pb.
Los amplicones del tamaño esperado correspondientes al menos a cuatro aislamientos representativos de cada uno de los cuatro departamentos bajo estudio, fueron purificados del gel mediante el kit QIAquick Gel Extraction, para realizar su secuenciación directa en ambos sentidos utilizando los mismos cebadores empleados en la Pcr y el Big Dye Terminator Cycle Sequencing Ready Reaction kit. Su corrido y separación se realizó en un secuenciador ABI Prism 3730xl.
A partir de las secuencias obtenidas con cada cebador, se construyeron las secuencias consenso mediante los programas bioinformáticos BioEdit 6.0.6 (Hall 1999) y Chromas Lite (Technelysium 2009). Posteriormente se confirmó su origen viral por comparación con las bases de datos moleculares con el programa BLAST (Altschul et al. 1990) y se obtuvieron secuencias de PMTV del GenBank de otras regiones del mundo, para realizar un análisis filogenético. Para esto, se generó un alineamiento mediante Clustal W (Larkin et al. 2007) del programa Bioedit 6.0.6. Los alineamientos fueron utilizados en una matriz de distancia genética utilizando el método de Máxima Verosimilitud basado en el modelo de Tamura-nei (1993) implementado en el programa MEGA v4.0 (Tamura et al. 2007). Para evaluar el soporte estadístico de cada agrupación, se realizó un análisis de Bootstrap con 1000 remuestreos (Felsenstein 1985).
Secuenciación del ARN 2 y ARN 3 de aislamientos colombianos de PMTV
Con el fin de obtener un mayor nivel de información sobre las características de los segmentos ARN 2 y ARN 3 del genoma de aislamientos colombianos de PMTV, se realizó un proceso de secuenciación con minado de cebadores, previamente reportados para la amplificación de diferentes regiones de dichos segmentos (Cuadro 1) y buscando que generaran traslape de secuencias para la construcción de un contig. Para esto se utilizaron dos cepas de PMTV de Antioquia (23T) y Boyacá (BS4T), seleccionadas gracias a la disponibilidad del material biológico necesario para su evaluación. Con cada cepa se realizaron por lo menos dos amplificaciones con cada cebador, de manera que las secuencias reportadas correspondieran al consenso para cada amplicón. Los procedimientos de extracción de ARN, PCR y secuenciación fueron similares a los descritos anteriormente, aunque las RT se realizaron con la enzima Transcriptasa Reversa Máxima, que ofrece mayores niveles de estabilidad térmica (hasta 65°C) y eficiencia que las transcriptasas reversas convencionales. en este caso las reacciones de rT consistieron de 9,5 µl de agua libre de nucleasas, 4 µl de buffer RT (5X), 1 µl de dNTPs (10 mM), 1 µl de cebador reverso (10 µM), 0,5 µl de inhibidor de ARNasas (40U/µl), 1 µl de Transcriptasa Reversa (200 U/µl) y 3 µl de ARN, para un volumen final de 20 µl. La incubación se realizó a 50°C por 30 min y la enzima se inactivó a 85°C por 5 min.
Las secuencias se editaron mediante el programa Chromas Lite, generándose los consensos con ambos cebadores con el programa Bioedit 6.0.6 y se procedió al ensamblaje de los contigs resultantes con el programa CAP3 (Huang y Madan 1999). Posteriormente, se verificó el marco de lectura correcto utilizando el portal bioinformático Expasy (Artimo et al. 2012) y se realizó su alineamiento con respecto a las secuencias de los dos segmentos genómicos de aislamientos cuyas secuencias completas se encontraban disponibles en genBank (DQ102381, DQ144451, AJ243719, AJ277556) (Sandgren et al. 2001, Cerovska et al. 2007).
Las secuencias de nucleótidos fueron traducidas y se compararon los porcentajes de identidad y número de posiciones variables a lo largo de las regiones bajo estudio y generándose árboles filogenéticos basados en los contigs obtenidos en la investigación, mediante el programa Mega v. 4.0, bajo los parámetros antes descritos.
Los números de accesión de las secuencias obtenidas en este trabajo corresponden a las series: JX889608-JX889611 y JX885612-JX885631.
Resultados y Discusión
Variabilidad genética de PMTV en Colombia
En total se obtuvieron 25 secuencias para la región amplificada de CP y 38 para TGB2. Estas correspondieron a por lo menos cuatro aislamientos por cada departamento bajo análisis (Cuadro 2). De esta forma se aumentaron las secuencias parciales de los genes CP y TGB2 de PMTV con respecto al trabajo reciente realizado por gil et al. (2011) en donde solo se obtuvieron cuatro y tres secuencias, para CP y TGB2; lo cual implica que las diferentes modificaciones realizadas a los procedimientos necesarios para el estudio de este virus, desde la forma de obtención de las muestras hasta la purificación de los amplicones, pasando por la eliminación de inhibidores de las polimerasas, resultaron altamente eficientes. Esta situación abre la posibilidad de plantear en el futuro un estudio de incidencia de PMTV basado en la detección del virus mediante RTPCR, con un muestreo estratificado en los diferentes pisos térmicos donde se cultiva la papa en Colombia y realizando comparaciones con respecto a los niveles de incidencia de Sss, de manera que sea posible determinar la dimensión del grado de dispersión de este virus en Colombia. Un trabajo similar fue realizado por montero-astúa et al. (2008) en costa rica pero utilizando pruebas de eLisa tanto para la detección de Sss como de PMTV. En este caso se encontró que el virus era más frecuentemente detectado en las regiones con mayores altitudes, donde las condiciones de alta humedad y bajas temperaturas aparentemente favorecen su infección. Sin embargo, es probable que la utilización de pruebas de ELISA, haya conducido a una subestimación de los niveles de incidencia detectados en dicho trabajo, pues la distribución errática del virus, su capacidad de movimiento célula-célula con ayuda de proteínas de movimiento codificadas por el TGB y la utilización de anticuerpos diseñados a partir de cepas del norte de Europa, pueden influir negativamente en la detección serológica del PMTV; aunque la problemática de la distribución del virus, también afecta la detección de técnicas como el RT-PCR (Gallo 2012).
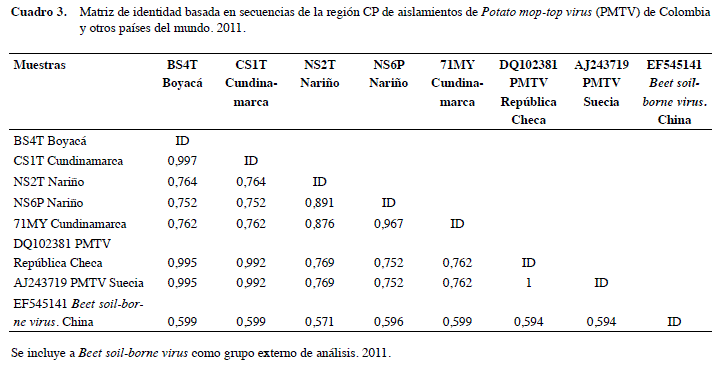
El análisis filogenético para CP incluyó 404 posiciones a partir de 46 secuencias, 15 de las cuales corresponden a aislamientos de referencia de europa, Asia y Norteamérica (Estados Unidos y Canadá), cuatro fueron previamente reportadas de aislamientos colombianos por gil et al. (2011), 25 fueron obtenidas en este estudio y las dos restantes corresponden al grupo externo de análisis (Beet soil-borne virus) y a una secuencia utilizada como control positivo obtenida de un kit comercial de ELISA para PMTV. El dendrograma presentó dos grupos soportados por valores de bootstrap del 100% (Figura 1). El primer clado agrupó todas las secuencias de referencia con 19 de los aislamientos colombianos obtenidos en este estudio y presentaron niveles de identidad superiores al 99% en todos los casos (Cuadro 3). Por otra parte, el segundo clado presentó exclusivamente secuencias de cepas colombianas, incluyendo seis de las obtenidas en este estudio y dos de las reportadas por gil et al. (2011). al interior de este clado, se presentaron mayores niveles de variación (11 a 13%) y su identidad con respecto a los miembros del primer clado fue de tan solo 76%. De manera interesante, en este clado no se ubicó ninguno de los aislamientos de PMTV obtenidos en Antioquia, a pesar de que el material de siembra de papa utilizado en este departamento, generalmente es adquirido en la sabana Cundiboyacense y Nariño, lugares en donde si se detectaron las variantes de PMTV. Por esto, dicho hallazgo debería ser considerado en estudios futuros, para evaluar si se trata de una situación aleatoria debido al tamaño de muestra o a factores no evaluados en este trabajo, como las características de los genotipos del vector Sss presentes en este departamento o a diferencias en las condiciones agroclimáticas propias de los municipios antioqueños cultivadores de papa, entre otros aspectos.
La secuencia de Beet soil-borne virus de China, utilizada como grupo externo de análisis, efectivamente se ubicó en posición externa a los dos clados y compartió niveles de identidad inferiores al 59% con respecto a los aislamientos de PmTV.
Estos resultados confirman los hallazgos de Vélez (2007) y gil et al. (2011) en referencia a la presencia de una variante viral de PMTV en Colombia, que incluso bajo los parámetros definidos por Adams et al. (2009), para la definición de especies al interior de la familia Virgaviridae (ej. cepas con identidades superiores a 80 y 90% a nivel de secuencias de CP y del genoma completo, respectivamente) podría representar una nueva especie de pomovirus. Si se considera que seis de las 25 secuencias de cP obtenidas en este trabajo correspondieron a dicha variante, es evidente que su presencia no es marginal en los cultivos de papa de Colombia, ya que para la muestra obtenida, aunque evidentemente sesgada por la colección de suelos y tejidos con presencia de estructuras de Sss, su proporción es del 23%.
Estos resultados difieren con respecto a lo encontrado en los estudios de variabilidad de PMTV con base en secuencias de CP de diferentes orígenes geográficos, por cuanto indistintamente se había encontrado que los niveles de identidad presentes en este gen superaban el 98% en todas las comparaciones establecidas. así por ejemplo, al comparar 28 secuencias de Finlandia, Letonia, Escocia, Dinamarca, República Checa y Suecia, casi todas eran idénticas, siendo tan solo detectado un 2% de variación entre las más distantes, lo cual representaba cambios en tan solo siete posiciones a nivel de la secuencia de aa de la proteína resultante (Latvala-Kirby et al. 2009). similarmente, Xu et al. (2004) reportaron que aislamientos de PMTV de Estados Unidos y Canadá eran idénticos y que estos a su vez tan solo diferían en máximo 3% de sus secuencias de cP con respecto a cepas de Perú (mayo et al. 1996), Escocia y Escandinavia (Reavy et al. 1997). Los mayores niveles de diversidad encontrados en este trabajo para aislamientos de PmTV, pueden deberse al hecho que la región Andina de Colombia hace parte del centro de origen de la papa, y es ampliamente aceptado que en estas zonas geográficas ocurren mayores niveles de variación no solo en los hospedantes, sino también en sus patógenos, dados los largos periodos de coevolución entre estos (Salazar 2006). De esta forma, de gran interés resultará emprender estudios que permitan evaluar las diferencias biológicas de dichas variantes de PMTV sobre distintas variedades de papa e incluso sobre hospedantes experimentales, debido a que sí existen diferencias de patogenicidad entre aislamientos de PMTV que presentan bajos niveles de variación (Nielsen y Nicolaisen 2003), es posible que dicho efecto sea aún mayor cuando se evalúen las variantes aquí reportadas.
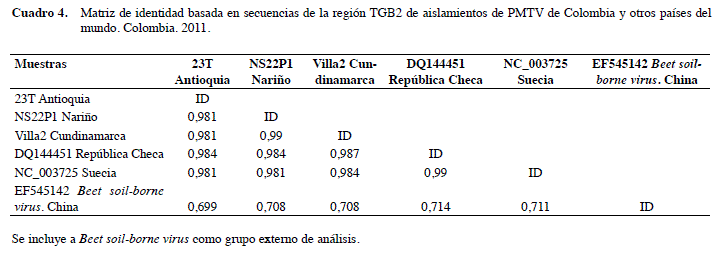
Con respecto al análisis filogenético basado en las secuencias de TGB2, se incluyeron 330 posiciones de 49 secuencias, siete de las cuales fueron de aislamientos de referencia de diferentes países y tres de colombia previamente analizadas (Gil et al. 2011). el dendrograma generado presentó un solo clado que incluyó todas las secuencias (Figura 2), las que compartieron niveles de identidad superiores a 98% (Cuadro 4).
Estos resultados son un indicativo de que la variación encontrada entre aislamientos colombianos con respecto a CP, no necesariamente se presentan a lo largo de los diferentes segmentos genómicos de PMTV, y específicamente no se ven reflejadas en las secuencias de TGB2; que en los pocos estudios donde se ha secuenciado, presenta niveles de identidad superiores a 94,8%. Incluso se ha encontrado que aislamientos de orígenes geográficos diferentes pueden compartir hasta el 100% de identidad en la secuencia de aa para este gen (ej. aislamiento Korneta-Nemilkov de República Checa y aislamientos 54-15 y 54-19 de Dinamarca) (Cerovska et al. 2003, 2007).
Los altos niveles de conservación de este gen entre aislamientos de PMTV, posibilitan que los cebadores utilizados para su amplificación sean una herramienta útil para el diagnóstico de este virus. así por ejemplo, Lambert et al. (2003) diseñaron los cebadores PMTV5 y PMTV7 dirigidos a amplificar una región de 646 pb comprendida entre TGB1 y TGB2 cuando reportaron por primera vez la presencia del PMTV en EEUU y posteriormente Xu et al. (2004), utilizaron la región que codifica para TGB2 para el diseño de los cebadores PMTVF4 y PMTVR4 en su estudio de evaluación de distribución de PMTV en EEUU y Canadá.
Secuenciación del ARN 2 y ARN 3 de aislamientos colombianos de PMTV
Para el ARN 2 se obtuvieron contigs de 2577 nt y 2584 para los aislamientos de Antioquia y Boyacá; mientras que para el ARN 3 los contigs tuvieron una extensión de 2617 y 2360 nt, respectivamente. Esto implica que para el ARN 2 se obtuvo un porcentaje de cobertura del 83% de este segmento genómico y del 87% para el ARN 3, en comparación con el aislamiento Sw de PMTV (AJ243719 y AJ277556) (Sandgren et al. 2001).
En términos de aa los contigs del arn2 incluyeron 150 de los aa de cP (posiciones 26 a 176) y 799 de CP-RT (posiciones 26 a 825). Para el ARN 3 se obtuvo la secuencia completa de aa de TGB1 (463 aa) y TGB2 (119 aa), así como 146 de 190 aa de TGB3 (posiciones 1 a 146) para el aislamiento de Boyacá y la totalidad de dicha proteína para la cepa de Antioquia. Las demás secuencias obtenidas corresponden a regiones UTR de los extremos 3´ para el ARN 2 y 5´ para el ARN 3.
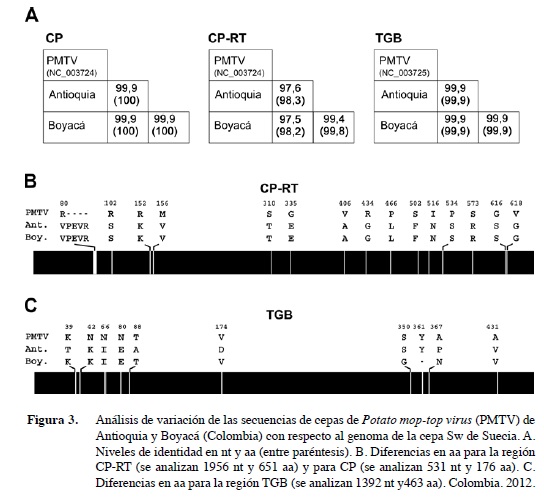
Con el fin de realizar un análisis de variación de las secuencias de las dos cepas obtenidas con respecto al genoma de la de PMTV de Suecia – Sw (Savenkov et al. 1999, Sandgren et al. 2001), se incluyeron 1956 nt (651 aa) para CP-RT, 531 nt (176 aa) para CP y 1392 (463 aa) para las tres regiones del TGB. En términos generales se encontraron muy altos niveles de identidad en las tres regiones analizadas, siendo la cP la más uniforme con 99% en la secuencia de nt y 100% en aa para las tres cepas. CP-RT presentó un nivel de identidad para nt de 99,4% entre las cepas de Antioquia y Boyacá y del 97,5 - 97,6% con respecto a la secuencia de referencia; mientras que estos valores fueron superiores al 98,2% cuando se comparó la secuencia de aa. Para TGB se encontró que todos los aislamientos compartieron un 99,9% de identidad en nt y aa (Figura 3). Las pocas diferencias a nivel de aa encontradas en este estudio, correspondieron a cambios en quince posiciones para el producto del ARN 2, siendo la más relevante una inserción de 4 aa (PEVR) a partir de la posición 81 de CP. Para el caso de TGB se encontraron diez cambios a lo largo de las tres proteínas analizadas, con la presencia de una deleción en la posición 361 en la cepa de Boyacá.
Por otra parte, el análisis filogenético realizado con base en 2569 nt para el ARN 2 con secuencias de referencia de los aislamientos Corneta-Nemilkov de República Checa (DQ102381) y Sw (AJ243719) de Suecia (Figura 4), arrojó un dendrograma con dos clados soportados por 100% de bootstrap, separándose los aislamientos de colombia de los de europa; sin embargo, los niveles de identidad entre ambos grupos fueron muy altos (>97,5%). Al calcular los niveles de diversidad (promedio del número de sustituciones de nucleótidos por cada sitio), para sitios sinónimos (dS) y no sinónimos (dNS), se encontró una diversidad muy baja, con tan solo 0,017 (se= 0,003) para dns y 0,009 (SE=0,002) para dS. La relación promedia entre dNS/ dS fue de 1,8, lo cual indica que las cepas de PMTV se encuentran bajo selección positiva para este componente del genoma. Para las cepas colombianas el valor de dNS/dS fue de 3, mientras que para las europeas fue de 1,2. La distancia genética basada en el método de Ki-mura-2 parámetros, para toda la población fue de 0,015.
El análisis filogenético realizado con base en 2008 nt para arn 3, con secuencias de referencia de los aislamientos Korneta-Nemilkov (DQ144451), 54-19 (AY353719) y 54-10 (AY426745) de Dinamarca, Sw (AJ277556) de Suecia y Todd (D30753) de Escocia (Figura 5) generó un dendrograma con dos clados principales de aislamientos europeos, mientras que los colombianos se presentaron en posición intermedia entre estos, en ramas soportadas por 100% de bootstrap. A pesar de dichas agrupaciones, los niveles de identidad entre todos los aislamientos fueron superiores a 96,7%. Al calcular los niveles de diversidad para sitios sinónimos (dS) y no sinónimos (dNS), se encontró una diversidad muy baja para todos los aislamientos, con tan solo 0,011 (se= 0,002) para dns y 0,044 (SE=0,006) para dS. La relación media entre dNS/dS fue de 0,25, lo cual indica que las cepas de PMTV se encuentran bajo selección negativa en dicho segmento de ARN. Para las cepas colombianas el valor de dNS/ dS fue de 0,19, mientras que para los aislamientos europeos fue de 0,39. La distancia genética basada en el método de Kimura -2 parámetros, para toda la población fue de 0,020 (SE 0,002).
Los bajos niveles de variación encontrados en los análisis de genomas de PMTV (Savenkov et al. 1999, Sandgren et al. 2001, Cerovska et al. 2007) y que nuevamente fueron inferidos en el presente estudio para las cepas del clado que representa PMTV sensu stricto, indican que este virus se encuentra bajo fuertes presiones de selección determinadas presumiblemente por las propias características de su genoma, el estrecho ámbito de hospedantes y su transmisión persistente y específica por parte de su vector Sss (Kirk 2008, Santala et al. 2010). Para el primer caso, la restricción selectiva se manifiesta por los bajos valores de dNS/dS calculados para dicho segmento y ocurre fundamentalmente al presentarse el ARN 2, dicha relación fue >1, lo cual sugiere una selección positiva, al presentarse menos restricciones para la fijación de mutaciones en los genes que codifican para CP y CP-RT. De otra parte, la presencia de un estrecho ámbito de hospedantes para el virus, que se restringe a algunas especies de las familias Solanaceae y Chenopodiaceae (Kirk 2008), sumada a su transmisión por Sss supone fuertes restricciones sobre los cambios en las diferentes proteínas del virus que interactúan con sus hospedantes y vectores, lo cual impide altas tasas de variación, tales como las que presentan otros virus de ARNss(+) como los potyvirus, que en general tienen amplios ámbitos de hospedantes y cuya transmisión por áfidos polífagos es del tipo no persistente (Hu et al. 2011).
De forma interesante, estudios recientes realizados por osorio et al. (2012), han encontrado que en Colombia se presenta un mayor nivel de variación con respecto a otros países, entre las poblaciones de sss, vector de PMTV, al registrarse además de los Tipos mundialmente reportados de este patógeno (I y II), un tercer Tipo (III), que presenta porcentajes de divergencia entre 2% y 5%, con respecto a los anteriores.
El resultado de las inserciones y diferentes sustituciones encontradas en este trabajo en los segmentos de ARN 2 y 3, requiere un tratamiento futuro detallado a partir de análisis de mutantes y de sus consecuencias sobre el ciclo infectivo viral. Igualmente, es imperativo estudiar el efecto de los dos genotipos virales detectados en este estudio sobre la producción y calidad del tubérculo semilla de papa en Colombia, de manera que los organismos de sanidad vegetal estatal, gremios de productores y agroindustrias, puedan valorar la necesidad de incluir la detección del PMTV en los programas de certificación de semilla y de mejoramiento genético de papa.
Agradecimientos
Esta investigación se realizó gracias al apoyo económico y técnico del Ministerio de Agricultura y Desarrollo Rural (proyecto 090-2007S4527-87-08), de la Universidad Nacional de Colombia, sede Medellín, Fedepapa y Fritolay. Se agradece a la Prof. Luz Estela Lagos, de la Universidad de Nariño por su apoyo con la colección de muestras en el Departamento de Nariño.
Literatura citada
Adams, MJ; Antoniw, JF; Kreuze, J. 2009. Virgaviridae: a new family of rod-shaped plant viruses. Archives of Virology 154:1967-1972. [ Links ]
Altschul, SF; Gish, W; Miller, W; Myers, EW; Lipman, DJ. 1990. Basic local alignment search tool. Journal of Molecular Biology 215:403-410. [ Links ]
Artimo, P; Jonnalagedda, M; Arnold, K; Baratin, D; Csardi, G; de Castro, E; Duvaud, S; Flegel, V; Portier, A; Gasteiger, E; Grosdidier, A; Hernandez, C; Loannidis, V; Kuznetsov, D; Liechti, R; Moretti, S; Mostaguir, K; Redaschi, N; Dossier, G; Xenarios, I; Stockinger, H. 2012. ExPASy: SIB bioinformatics resource portal, Nucleic Acids Research 40:597-603. [ Links ]
Cerovska, N; Moravec, T; Rosecka, P; Filigarova, M; Pecenkova, T. 2003. Nucleotide sequences of coat protein coding regions of six potato mop-top virus isolates. Acta Virologica 47:37-40. [ Links ]
Cerovska, N; Pecenkova, M; Foligarova, M; Dedic, P. 2007. Sequence analysis of the Czech Potato mop-top virus (PMTV) isolate Korneta-Nemilkov. Folia Microbiologica 52:61-64. [ Links ]
Cowan, GH; Torrance, L; Reavy, B.1997. Detection of Potato mop-top virus capsid readthrough protein in virus particles. Journal of General Virology 78:1779-1783. [ Links ]
Davey, T. 2009. The importance of Potato mop-top virus (PMTV) in Scottish seed potatoes (PhD Thesis). Heriot Watt University & Science and Advice for Scottish Agriculture. Scotland. 178 p. [ Links ]
Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783-791. [ Links ]
Gallo, Y. 2012. Generación de antígenos derivados de la proteína de la cápside de PVY, TaLMV y PMTV, para la producción de anticuerpos útiles en el desarrollo de pruebas serológicas. Tesis de Maestría en Bioquímica. Facultad de Medicina, Universidad Nacional de Colombia, sede Bogotá. Colombia. 148 p. [ Links ]
Gil, JF; Gutiérrez, P; Cotes, JM; González, EP; Marín, M. 2011. Caracterización genotípica de aislamientos colombianos del Potato mop-top virus (PmTV, Pomovirus). Actualidades Biológicas 33:69-84. [ Links ]
Guerrero, O. 2000. La roña o sarna polvosa en el Departamento de Nariño. Papas colombianas con el mejor entorno ambiental. 2 ed. Bogotá, Fedepapa, Colombia. p. 127-129. [ Links ]
Hall, TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41:95-98. [ Links ]
Harrison, BD; Jones, RAC. 1970. Host range and some properties of Potato mop-top virus. Annals Applied Biology 65:393-402. [ Links ]
Holdridge, LR. 1987. Ecología basada en zonas de vida. Trad. Humberto Jiménez Saa. IICA. San José, Costa Rica. 216 p. [ Links ]
Hu, X; Karasev, AV; Brown, CJ; Lorenzen, JH. 2009. Sequence characteristics of potato virus Y recombinants. Journal of General Virology 90:3033-3041. [ Links ]
Huang, X; Madan, A. 1999. CAP3: A DNA sequence assem-bly program. Genome Research 9:868-877. [ Links ]
Jaramillo, S; Botero, JM. 2007. Respuesta de diferentes poblaciones de Spongospora subterranea f. sp. subterranea a la rotación entre dos variedades de papa (Solanum tuberosum ssp. andigena). Revista Facultad Nacional de Agronomía Medellín 60:3859-3876. [ Links ]
Jones, RAC; Harrison, BD. 1972. Ecological studies on Potato mop-top virus in Scotland. Annals Applied Biology 71:47-57. [ Links ]
Kirk, HG. 2008. Mop-top virus, relationship to its vector. American Journal of Potato Research 85:261-265. [ Links ]
Lambert, DH; Levy, L; Mavrodieva, VA; Johnson, SB; Babcock, MJ; Vayda, ME. 2003. First report of Potato mop-top virus on potato from the United States. Plant Disease 87:872. [ Links ]
Larkin, M; Blackshields, G; Brown, NP; Chenna, R; McGettigan, PA; McWilliam, H; Valentin, F; Wallace, IM; Wilm, A; López, R; Thompson, JD; Gibson, TJ; Higgins, DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947-2948. [ Links ]
Latvala-Kilby, S; Aura, JM; Pupola, N; Hannukkala, A; Valkonen, JPT. 2009. Detection of Potato mop-top virus in potato tubers and sprouts: combinations of rna2 and RNA3 variants and incidence of symptomless infections. Phytopathology 99:519-531. [ Links ]
Lim, HS; Bragg, JN; Ganesan, U; Ruzin, S; Schichnes, D; Lee, MY; Vaira, AM; Ryu, KH; Hammond, J; Jackson, AO. 2009. Subcellular localization of the Barley stripe mosaic virus triple gene block proteins. Journal Virology 83:9432-9448. [ Links ]
MacKenzie, DJ. 1996. Detection of Potato Mop-top virus in leaf or tuber tissue by reverse transcription-polymerase chain reaction. Document CPHBT96K03, Centre for Plant Health, Agriculture and Agri-Food Canada. Sidney, B.C., Canadá. 7 p. [ Links ]
Mayo, MA; Torrance, L; Cowan, G; Jolly, CA; Macintosh, SM; Orrego, R; Barrera, C; Salazar, LF. 1996. Conservation of coat protein sequence among isolates of Potato mop-top virus from Scotland and Peru. Archives of Virology 141:1115-1121. [ Links ]
Montero-Astúa, M; Vasquéz, V; Turechek, WW; Merz, U; Rivera, C. 2008. Incidence, distribution, and association of Spongospora subterranea and Potato mop-top virus in Costa Rica. Plant Disease 92:1171-1176. [ Links ]
Mumford, RA; Walsh, K; Barker, I; Boonham, N. 2000. Detection of Potato mop top virus and Tobacco rattle virus using a multiplex real-time fluorescent reverse transcription polymerase chain reaction assay. Phytopathology 90:448-453. [ Links ]
Nakayama, T; Maoka, T; Hataya, T; Shimizu, M; Fuwa, H; Tsuda, S; Motoyuki, M. 2010. Diagnosis of Potato mop-top virus in soil using bait plant bioassay and RT-PCR-microplate hybridization. American Journal of Potato Research 87:218-225. [ Links ]
Nielsen, SL; Nicolaisen, M. 2003. Identification of two nucleotide sequence sub-groups within Potato mop-top virus. Archives of Virology 148:381-388. [ Links ]
Osorio, I; Orozco, M; Gutiérrez, P; González, E; Marín, M. 2012. Variabilidad genética de Spongospora subterranea f.sp. subterranea en Colombia. Bioagro 24:151-162. [ Links ]
Reavy, B; Sandgren, M; Barker, H; Heino, P; Oxelfelt, P. 1997. A coat protein transgene from a Scottish isolate of Potato mop-top virus mediates strong resistance against Scandinavian isolates which have similar coat protein genes. European Journal of Plant Pathology 103:829-834. [ Links ]
Salazar, LF. 2006. Emerging and re-emerging potato diseases in the Andes. Potato Research 49:43-47. [ Links ]
Sandgren, M; Savenkov, E; Valkonen, JP. 2001. The readthrough region of Potato mop-top virus (PmTV) coat protein enconding RNA, the second largest RNA of PMTV genome, undergoes structural changes in naturally infected and experimentally inoculated plants. Archives of Virology 146:467-477. [ Links ]
Santala, J; Samuilova, O; Hannukkala, A; Latvala, S; Kortemaa, H; Beuch, U; Kvarnheden, A; Persson, P; Topp, K; Ørstad, K; Spetz, C; Nielsen, SL; Kirk, HG; Budziszewska, M; Wieczorek, P; Obrępalska-Stęplowska, A; Pospieszny, H; Kryszczuk, A; Sztangret-Wiśniewska, J; Yin, Z; Chrzanowska, M; Zimnoch-Guzowska, E; Jackeviciene, E; Taluntytė, L; Pūpola, N; Mihailova, J; Lielmane, I; Järvekülg, L; Kotkas, K; Rogozina, E; Sozonov, A; Tikhonovich, I; Horn, P; Broer, I; Kuusiene, S; Staniulis, J; Uth, JG; Adam, G; Valkonen, JPT. 2010. Detection, distribution and control of Potato mop-top virus, a soil-borne virus, in northern Europe. Annals of Applied Biology 157:163-178. [ Links ]
Savenkov, EI; Sandgren, MY; Valkonen, JPT. 1999. Complete sequence of RNA 1 and the presence of tRNA-like structures in all rnas of Potato mop-top virus, genus Pomovirus. Journal of General Virology 80:2779-2784. [ Links ]
Savenkov, EI; Germundsson, A; Zamyathin, AA; Sandgren, M; Valkonen, JPT. 2003. Potato mop-top virus: The coat protein-encoding RNA and the gene for cysteinerich protein are dispensable for systemic virus movement in Nicotiana benthamiana. Journal of General Virology 84:1001-1005. [ Links ]
Tamura, K; Nei, M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10:512-526. [ Links ]
Tamura, K; Dudley, J; Nei, M; Kumar, S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution 24:1596-1599. [ Links ]
Technelysium. 2009. Chromas. 2.0 (en línea). Consultado 15 febrero de 2009. Disponible en http://www.technelysium.com.au/chromas_lite.html. [ Links ]
Tenorio, J; Franco, Y; Chuquillanqui, C; Owens, RA; Salazar, LF. 2006. Reaction of potato varieties to Potato mop-top virus infection in the Andes. American Journal of Potato Research 83:423-31. [ Links ]
Torrance, L; Cowan, G. H; Sokmen, MA; Reavy, B. 1999. A naturally occurring deleted form of RNA 2 of Potato mop-top virus. Journal of General Virology 80:2211-2215. [ Links ]
Vélez, PB. 2007. Detección e identificación del Potato moptop virus (PMTV) en áreas de producción de papa donde se encuentra Spongospora subterranea en dos departamentos de colombia. Tesis de Maestría en Ciencias Agrarias. Facultad de Agronomía, Universidad Nacional de Colombia, sede Bogotá. Colombia. 115 p. [ Links ]
Xu, H; Dehaan, TL; De Boer, SH. 2004. Detection and confirmation of Potato mop-top virus in potatoes produced in the United States and Canada. Plant Disease 88:363-367. [ Links ]
*Correspondencia para:
Inés Osorio-Giraldo. Laboratorio de Biología Celular y Molecular y Laboratorio de Microbiología Industrial, Facultad de Ciencias, Universidad Nacional de Colombia - Sede Medellín, Colombia. Calle 59A No 63 - 20, Medellín, Colombia. inesosorio@gmail.com.
Pablo Gutiérrez-Sánchez. Laboratorio de Biología Celular y Molecular y Laboratorio de Microbiología Industrial, Facultad de Ciencias, Universidad Nacional de Colombia - Sede Medellín, Colombia. Calle 59A No 63 - 20, Medellín, Colombia. paguties@unal.edu.co.
Mauricio Marín-Montoya. Laboratorio de Biología Celular y Molecular y Laboratorio de Microbiología Industrial, Facultad de Ciencias, Universidad Nacional de Colombia - Sede Medellín, Colombia. Calle 59A No 63 - 20, Medellín, Colombia. mamarinm@unal.edu.co.
1. Parte de la tesis de maestría de la primera autora, desarrollada en el marco del proyecto 090-2007S4527-87-08, financiado por Ministerio de Agricultura y Desarrollo Rural, Colombia.
2. Laboratorio de Biología Celular y Molecular y Laboratorio de Microbiología Industrial, Facultad de Ciencias, Universidad Nacional de Colombia - Sede Medellín, Colombia. Calle 59A No 63 - 20, Medellín, Colombia. inesosorio@gmail.com, paguties@unal.edu.co, mamarinm@unal.edu.co (autor para correspondencia).
Inés Osorio-Giraldo. Laboratorio de Biología Celular y Molecular y Laboratorio de Microbiología Industrial, Facultad de Ciencias, Universidad Nacional de Colombia - Sede Medellín, Colombia. Calle 59A No 63 - 20, Medellín, Colombia. inesosorio@gmail.com.
Pablo Gutiérrez-Sánchez. Laboratorio de Biología Celular y Molecular y Laboratorio de Microbiología Industrial, Facultad de Ciencias, Universidad Nacional de Colombia - Sede Medellín, Colombia. Calle 59A No 63 - 20, Medellín, Colombia. paguties@unal.edu.co.
Mauricio Marín-Montoya. Laboratorio de Biología Celular y Molecular y Laboratorio de Microbiología Industrial, Facultad de Ciencias, Universidad Nacional de Colombia - Sede Medellín, Colombia. Calle 59A No 63 - 20, Medellín, Colombia. mamarinm@unal.edu.co.
1. Parte de la tesis de maestría de la primera autora, desarrollada en el marco del proyecto 090-2007S4527-87-08, financiado por Ministerio de Agricultura y Desarrollo Rural, Colombia.
Recibido: 28 de febrero, 2012. Aceptado: 1 de abril, 2013.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}