










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Cardiología
Print version ISSN 1409-4142
Rev. costarric. cardiol vol.14 n.1-2 San José Jan./Dec. 2012
Resumen de guías europeas
Hipertensión arterial pulmonar
Guías de manejo
Dra. Ileana Bolaños Campos, Dr. Randall Guadamuz Vásquez, Dra. Marlen Jiménez Carro, Dra. Betty Rocha Contreras, Dr. Eduardo Sáenz Madrigal & Dr. Carlos Salazar Vargas*+
Estas guías son un resumen de las Guías Europeas publicadas en la Revista Española de Cardiología 2009, 62:1464.e, disponible en versión electrónica: www.revesp.cardiol.org
En nombre de los pacientes, futuros beneficiarios, agradecemos profundamente a la compañía Bayer por el apoyo constante a esta iniciativa
Abreviaturas y acrónimos
AIR: estudio aleatorizado del iloprost en aerosol (del inglés, Aerolized Iloprost Randomized study).
ALPHABET: ensayo europeo de hipertensión arterial pulmonar y beraprost (del inglés, Arterial Pulmonary Hypertension and Beraprost
European Trial).
AP: arteria pulmonar.
ARE: antagonista del receptor de la endotelina.
BCC: bloqueador de los canales de calcio.
BENEFIT: efectos del bosentán en formas de hipertensión pulmonar tromboembólica crónica inoperable (del inglés, Bosentan Effects iN iNopErable Forms of chronic Thromboembolic pulmonary hypertension).
BNP: péptido natriurético cerebral (del inglés,brain natriuretic peptide).
BREATHE: ensayo aleatorizado de bosentán sobre terapia con antagonista de la endotelina (del inglés, bosentan randomized trial of endothelin antagonist therapy).
CCD: cateterismo cardiaco derecho.
CF de la OMS: clase funcional de la Organización Mundial de la Salud.
CIA: comunicación interauricular.
COMBI: terapia de combinación de bosentán e iloprost en aerosol en la hipertensión arterial pulmonar idiopática (del inglés, COMbination therapy of Bosentan and aerolised Iloprost in idiopathic pulmonary arterial hypertension).
DLCO: capacidad de difusión del monóxido de carbono
EAP: endarterectomía de la arteria pulmonar.
EARLY: ensayo sobre el antagonista de la endotelina en pacientes con hipertensión arterial pulmonar levemente sintomáticos (del inglés, Endothelin Antagonist tRial in mildLY symptomatic pulmonary arterial hypertension patients).
ECC: enfermedad cardiaca congénita.
ECDA: ensayo controlado con distribución aleatoria.
ECG: electrocardiograma
EPOC: enfermedad pulmonar obstructiva crónica.
ESPAT: excursión sistólica del plano anular tricúspide. ETC: enfermedad del tejido conectivo.
ETC: enfermedad del tejido conectivo.
ETT: eco cardiografía tránsito radica.
EVOP: enfermedad veno-oclusiva pulmonar.
GC: gasto cardiaco.
GPT: gradiente de presión transpulmonar (PAP media-PEP media).
HAP: hipertensión arterial pulmonar.
HAPA: hipertensión arterial pulmonar asociada.
HAPI: hipertensión arterial pulmonar idiopática.
HP: hipertensión pulmonar.
HPTC: hipertensión pulmonar tromboembólica crónica.
HPTEC: hipertensión pulmonar trombo embolica.
I.V.: intravenoso.
IC: índice cardiaco.
IFD5: inhibidores de la fosfodiesterasa
NO: óxido nítrico.
NT-proBNP: fracción N-terminal del propéptido natriurético cerebral (del inglés, N-terminal brain natriuretic peptide).
PACES: estudio de la combinación de epoprostenol y sildenafilo en la hipertensión arterial pulmonar (del inglés, Pulmonary Arterial hypertension Combination study of Epoprostenol and Sildenafil).
PAD: presión auricular derecha.
PAP: presión arterial pulmonar.
PEP: presión de enclavamiento pulmonar.
PHIRST: ensayo sobre la hipertensión arterial pulmonar y respuesta al tadalafilo (del inglés, Pulmonary arterial Hypertension and ReSponse to Tadalafil trial).
PM6M: prueba de marcha de 6 min.
RNI: razón normalizada internacional.
RVP: resistencia vascular pulmonar.
SAB: septostomía auricular con balón.
STEP: ensayo piloto sobre la seguridad y eficacia del iloprost inhalado en combinación con bosentán para la evaluación en la hipertensión arterial pulmonar (del inglés, Safety and pilot efficacy Trial of inhalated iloprost in combination with bosentan for Evaluation in Pulmonary arterial hypertension).
STRIDE: sitaxentán para aliviar el ejercicio deficiente (del inglés, Sitaxentan To Relieve ImpaireD Exercise).
SUPER: ensayo sobre el uso del sildenafilo en la hipertensión arterial pulmonar (del inglés, Sildenafil Use in Pulmonary arterial
hypERtension). t.i.d.: 3 veces al día (del latín, ter in die).
TC: tomografía computarizada.
TRIUMPH: treprostinil sódico inhalado en pacientes con hipertensión arterial pulmonar grave (del inglés, inhaled TReprostinil sodIUM in patients with severe Pulmonary arterial Hypertension).
VD: ventrículo derecho.
VI: ventrículo izquierdo.
VIH: virus de la inmunodeficiencia humana.
Se valoró el nivel de evidencia y la fuerza de la recomendación según escalas conocidas. (Cuadros 1 y 2)
Hipertension pulmonar
Introducción
Definiciones
La hipertensión pulmonar (HP) es una enfermedad hemodinámica y patofisiológica definida como un aumento de la presión arterial pulmonar (PAP) media ≥ 25 mmHg en reposo, medida durante el cateterismo cardiaco derecho (Cuadro 3); desde el punto de vista hemodinámico se divide en HP precapilar y postcapilar.
Clasificación clínica
La HP puede presentarse en múltiples enfermedades clínicas, que han sido agrupadas por la OMS en 6 diferentes grupos. Según la clasificación actual de Dana Point 2008 (Cuadro 4). En esta nueva clasificación se hicieron cambios con respecto a la previa que son importantes de resaltar:
• Grupo 1 HAP: El término usado de HAP familiar es sustituido por HAP heredable, por existir mutaciones específicas en casos esporádicos sin antecedentes familiares. Dentro de éstas se encuentran la HAP idiopática esporádica (HAPI).
El grupo de HAP asociada a (HAPA) (Cuadro 4), incluye enfermedades con representación clínica e histológica similar, inclusive lesiones plexiformes similar a las de HAPI; en ella se incluye la esquistosomiasis, la anemia hemolítica crónica como la anemia de células falciformes, la talasemia, la esferocitosis heridataria, la estomatocitosis, y la anemia microangiopática; también dentro de la HAPA se incluyen las cardiopatías congénitas cuya clasificación clínica se muestra en la Cuadro 6
y su clasificación anatomo-patofisiológica en la Cuadro 6.
• Grupo 1’ Incluye Enfermedad veno-oclusiva pulmonar y/o hemangiomatosis pulmonar. Son enfermedades difíciles de clasificar comparten algunas características con HAPI pero con diferencias importantes. Son una categoría distinta pero no totalmente separada del grupo 1 HAP.
Patología de la HP
Los hallazgos patológicos en la hipertensión pulmonar se pueden resumir en:
GRUPO 1’: Incluye la enfermedad veno oclusiva pulmonar, afecta las venas septales y las vénulas preseptales con lesiones fibróticas oclusivas, y afecta arterias pulmonares distales.
GRUPO 2: causada por cardiopatía izquierda., aumento de tamaño y ensanchamiento de venas pulmonares
GRUPO 3, HP causada por hipoxemia: hipertrofia medial y proliferación obstructiva de la íntima de las arterias pulmonares distales.
GRUPO 4. HPTC: trombos organizados adheridos a las paredes arteriales y en las zonas no ocluídas puede desarrollarse una arteriopatía
indistinguible de la HAP, incluyendo lesiones plexiformes.
GRUPO 5. HP con mecanismos poco claros o multifactoriales: se incluyen enfermedades heterogéneas con cuadros patológicos de etiología poco clara o multifactorial.
Patobiología de la HP
GRUPO 5. HP con mecanismos poco claros o multifactoriales.
Genética, epidemiología y factores de riesgo de HP
No hay datos comparativos sobre la prevalencia de los diferentes grupos de HP, pero un estudio que utilizó una definición de PAP sistólica de >40 mmHg reportó que 79% eran del grupo 2 (cardiopatía izda.), 10% eran del grupo 3 (enfermedades pulmonares), 4% del grupo 1 (HAP), <1% del grupo 4 y en 7%, no se pudo definir el diagnóstico.
Grupo 1. La prevalencia de HAP y la de HAPI es de 15 y de 5.9 casos/ millón de población adulta. Cuando la HAP se desarrolla en un contexto familiar existen mutaciones de línea germinal en el receptor de proteínas morfogenéticas óseas tipo 2 en el 70% de los casos.
Entre diferentes funciones biológicas, estos polipéptidos participan en el control de la proli-feración celular vascular. Se han identificado mutaciones de otros receptores para estas sustancias, como la cinasa tipo 1 similar a los receptores de activina y la endoglina, en su mayoría en pacientes con HAP con antece-dentes familiares o personales de telangiectasia hemorrágica hereditaria (síndrome de Rendu-Osler-Weber).
Se han identificado factores de riesgo para HP, que se clasifican en definitivos, probables, posibles o improbables. Definitivos, como en el caso de los inhibidores del apetito en la década de los sesenta, probable es cuando un estudio de casoscontroles de un único centro demostró asociación. Los niveles de riesgo de diferentes fármacos y toxinas aparecen en la Cuadro 7.
Grupo 4. HPTC: Reportes recientes indican que la prevalencia de la HPTC es del 3,8% en subrevivientes de embolias pulmonares agudas, la mayoría cree que la incidencia de la HPTC tras una embolia pulmonar aguda es del 0,5-2%. La HPTC pude encontrarse en pacientes sin ningún episodio clínico anterior de embolia pulmonar aguda o trombosis venosa profunda (hasta el 50% en diferentes series).
Grupo 5. HP con mecanismos poco claros o multifactoriales: la heterogeneidad de este grupo impide una descripción adecuada de la genética, la epidemiología y los factores de riesgo en esta guía de práctica clínica.
Hipertensión arterial pulmonar (grupo 1)
El término hipertensión arterial pulmonar (HAP) (grupo 1) se utiliza para la enfermedad clínica caracterizada por la presencia de HP precapilar (Cuadro 3) en ausencia de otras causas de HP precapilar y por tanto es un diagnóstico de exclusión.
HAP incluye un grupo heterogéneo de enfermedades (Cuadro 4), que comparten una similar presentación clínica y hemodinámica, y cambios patológicos en la microcirculación pulmonar virtualmente idénticos. La HP produce un aumento en la RVP que produce sobrecarga del ventrículo derecho (VD), hipertrofia y dilatación, del mismo y finalmente su fracaso y la muerte.
Presentación clínica
La alta sospecha clínica del médico es primordial para poder hacer el diagnóstico ya que los síntomas son inespecíficos. El síntoma más común es la disnea de esfuerzo. Otros síntomas son: fatiga, debilidad, dolor torácico y síncope.
Al examen físico se encuentran: elevación paraesternal izquierda, un componente pulmonar del 2do ruido cardiaco acentuado(S2), soplo pansistólico de regurgitación tricuspídea, soplo diastólico de insuficiencia pulmonar y un 3er sonido del VD. Pacientes con enfermedad
más avanzada pueden presentar ingurgitación yugular, hepatomegalia, edema periférico, ascitis y extremidades frías.
El examen físico puede aportar datos que sugieren la causa de la HP, por ejemplo la presencia de telangiectasias, ulceración digital y esclerodactilia en la escleroderma y los crépitos pueden indicar enfermedad pulmonar. Si se encuentra acropaquia digital en HAPI, deberían buscarse diagnósticos alternativos, como CC o EVOP
Electrocardiograma
El ECG puede proporcionar evidencia que respalde la HP: hipertrofia y sobrecarga del VD y dilatación auricular derecha. La hipertrofia del VD se encuentra en el 87% de los pacientes con HAPI y la desviación del eje hacia la derecha en el 79%. La ausencia de estos no excluye la presencia de HP. La sensibilidad (55%) y la especificidad (70%) del ECG son insuficientes para convertirse en una herramienta de exploración para detectar una HP.
Las arritmias ventriculares son raras. Las arritmias supraventriculares pueden estar presentes en estados avanzados, en particular el aleteo (flutter) auricular y la fibrilación auricular.
Radiografía torácica
Es anormal en el momento del diagnóstico en el 90% de los pacientes con HAPI. Los hallazgos incluyen: aumento del tamaño del tronco principal de la arteria pulmonar y sus ramas principales, amputación brusca de la vascularización periférica y cámaras derechas agrandadas en casos avanzados. La radiografía permite excluir enfermedades pulmonares asociadas (grupo 3) (Cuadro 4) y la hipertensión venosa pulmonar causada por cardiopatía izquierda (grupo 2). En general, el grado de la HP no está en correlación con el de las anomalías radiográficas.
La alta sospecha clínica del médico es primordial para poder hacer el diagnóstico ya que los síntomas son inespecíficos. El síntoma más común es la disnea de esfuerzo. Otros síntomas son: fatiga, debilidad, dolor torácico y síncope.
Al examen físico se encuentran: elevación paraesternal izquierda, un componente pulmonar del 2do ruido cardiaco acentuado(S2), soplo pansistólico de regurgitación tricuspídea, soplo diastólico de insuficiencia pulmonar y un 3er sonido del VD. Pacientes con enfermedad
más avanzada pueden presentar ingurgitación yugular, hepatomegalia, edema periférico, ascitis y extremidades frías.
El examen físico puede aportar datos que sugieren la causa de la HP, por ejemplo la presencia de telangiectasias, ulceración digital y esclerodactilia en la escleroderma y los crépitos pueden indicar enfermedad pulmonar. Si se encuentra acropaquia digital en HAPI, deberían buscarse diagnósticos alternativos, como CC o EVOP.
Pruebas de función pulmonar y análisis de gases en sangre arterial
Ecocardiografía
La ecocardiografía transtorácica debe realizarse siempre que se sospecha una HP.
El cálculo de la PAP se basa en la velocidad pico del flujo de regurgitación tricuspídea. La ecuación simplificada de Bernoulli describe la relación de la velocidad de regurgitación tricuspídea y el gradiente depresión pico de regurgitación tricuspídea = 4 Å~ (velocidad de regurgitación tricuspídea). Esta permite calcular la presión sistólica de la AP teniendo en cuenta la presión auricular derecha: la presión sistólica de la AP = gradiente de presión de regurgitación tricuspídea + presión auricular derecha calculada. La presión auricular derecha puede calcularse con el diámetro y la variación respiratoria de la vena cava inferior, aunque a menudo se asume un valor fijo de 5 o 10 mmHg. Cuando resulta difícil medir la velocidad pico de regurgitación tricuspídea (regurgitación tricuspídea muy leve/leve), la utilización de ecocardiografía de contraste (p. ej., solución salina agitada) aumenta de forma significativa la señal Doppler, permitiendo una medición adecuada de la velocidad pico de regurgitación tricuspídea.
De la misma manera, habría que considerar los gradientes sistólicos potenciales entre el VD y la AP.
Teóricamente, el cálculo de la PAP media a partir de la presión sistólica de la AP es posible (PAP media = 0,61 x presión sistólica de la AP + 2 mmHg). Esto podría permitir la utilización de medidas Doppler, empleando una definición establecida de la HP como PAP media ≥ 25 mmHg.
En aquellos con regurgitación tricuspídea grave, el uso de la ecuación de Bernoulli simplificada puede conducir a la subestimación de la presión sistólica de la AP. También son comunes las sobrestimaciones de la presión sistólica de la AP en > 10 mmHg.
Por ello, la HP no puede definirse con precisión por el valor de corte de la presión sistólica de la AP según el método Doppler.
Siempre deberían considerarse otras variables ecocardiográficas que puedan levantar o reforzar sospechas de HP independientemente de la velocidad de regurgitación tricuspídea. Éstas incluyen:
1. Un aumento de la velocidad de regurgitación de la válvula pulmonar
2. Una breve aceleración del tiempo de eyección del VD hacia la AP.
3. Dilatación de las cavidades derechas del corazón.
4. Forma y función anómalas del tabique interventricular.
5. Aumento en el grosor parietal del VD, dilatación de AD, y dilatación de la AP principal.
Este grupo de trabajo propone criterios para detectar la HP, con base en la velocidad pico de regurgitación tricuspídea y en la presión sistólica de la AP, calculada según Doppler en reposo suponiendo una presión atrial derecha normal de 5 mmHg y variables ecocardiográficas adicionales. Cuadro 8.
Gamagrafía pulmonar de ventilación /perfusión
Este estudio debe realizarse para descartar enfermedad tromboembólica. Es más sensible que la TC. Un estudio de probabilidad normal o baja excluye HPTC, con sensibilidad del 90-100% y especificidad del 94-100%. En pacientes con HAPI suele ser normal o pueden encontrarse defectos de perfusión de aspecto moteado, pero no segmentarios.
Tomografía axial de alta resolución
La TC sirve para descartar enfermedad pulmonar intersticial y cuando hay sospecha de EVOP o HCP.
La angiografía por TC se utiliza en la evaluación de los pacientes con HPTEC.
Arteriografía pulmonar
La arteriografía pulmonar convencional se utiliza para determinar en pacientes con HPTEC si son tributarios de EAP y puede ser de utilidad en malformaciones arteriovenosas pulmonares. Imágenes de resonancia magnética. Sirven para la valoración del tamaño, morfología y función del ventrículo derecho y permiten la valoración no invasiva del flujo sanguíneo (volumen latido, GC, distensibilidad de la AP, masa del VD) en el seguimiento de los pacientes. La disminución del volumen latido, el aumento del volumen diastólico final del VD y un descenso del volumen diastólico final de VI son de mal pronóstico. El aumento del volumen diastólico final del VD indica fracaso de esta cámara.
Análisis de sangre e inmunología
Se realizan pruebas de rutina bioquímica, hematológica y de función tiroidea en todos los pacientes. Las serológicas específicas sirven para ETC, VIH, y hepatitis. La esclerosis sistémica es la más importante de las ETC ya que tiene una alta prevalencia de HAP. En HPTC debe descartarse trombofilia realizando anticuerpos antifosfolípidos, los anticoagulantes lúpicos y los anticuerpos anticardiolipinas.
La prueba de VIH es obligatoria. Si hay datos de clínicos de hepatopatía se realizan pruebas de función hepática y serología para hepatitis.
Ecografía abdominal
Este estudio puede identificar cirrosis e hipertensión portal.
ECateterismo cardiaco derecho (CCD)
El diagnóstico final de HAP se realiza mediante CCD. También sirve para valorar la gravedad del deterioro hemodinámico y analizar la vasorreactividad de la circulación pulmonar. Se buscan la PAP (sistólica, diastólica y media), presión atrial derecha, PEP y presión de VD. Se determina el GC, las saturaciones de oxígeno de la vena cava superior, de la AP y de la sangre arterial sistémica. Se calculan las RVP. Una PEP> 15 mmHg excluye HAP precapilar.
Las variables hemodinámicas que deben ser efectuadas durante cateterismo cardiaco derecho:
1. Registro de la presión enclavada pulmonar (en cuña), de la presión sistólica – diastólica – y media de arteria pulmonar.
2. Registro de la presión en retiro, de tronco AP – VD - AD
3. EL gasto cardiaco debe medirse por triplicado ya sea termodilución o Fick , este último obligatorio en casos de shunt.
4. Saturación de Oxigeno de vena cava superior, de la arteria pulmonar, y sangre arterial sistémica.
5. Medición de las resistencias vasculares pulmonares.
6. Shunts y lesiones valvulares deben de ser verificados evaluados o excluidos. Angiografía coronaria debe de ser efectuada en los casos específicos.
Prueba de vasorreactividad
Las pruebas de vasorreactividad identifican los pacientes que se pueden beneficiar de una terapia a largo plazo con BCC.
Deben usarse vasodilatadores de acción inmediata, segura y fácil de administrar cuyos efectos sistémicos sean limitados o nulos. El agente más usado en pruebas agudas es el NO (Cuadro 9), aunque el epoprostenol I.V. y la adenosina son alternativas pero pueden generar efectos sistémicos. (Cuadro 9)
No deben usarse los BCC orales o I.V y tampoco existe evidencia para la utilización de Sildenafilo. Se define la respuesta aguda positiva aquella en la que la PAP media cae ≥ 10 mmHg para llegar a una PAP media de ≤ 40 mmHg con GC invariable o aumentado.
Los pacientes que responden positivamente en agudo, tienen más probablidades de mostrar una respuesta constante a tratamientos de larga duración y de altas dosis de BCC y son los únicos que pueden recibir este tratamiento de manera segura. La utilidad de las pruebas vasorreactivas agudas y del tratamiento de larga duración con BCC en pacientes con otros tipos de HAP no está tan clara como en la HAPI.
En la Cuadro 10 están las recomendaciones para las pruebas de CCD y pruebas de vasorreactividad.
Algoritmo diagnóstico
(Ver figura 1)
Si la valoración no invasiva es compatible con HP, el siguiente paso es descartar las causas más comunes que son las enfermedades del grupo 2 y 3 con los resultados del ecocardiograma transtorácico, pruebas de función pulmonar (oximetría nocturna, si fuera necesaria), gases arteriales y la TC de alta resolución torácica. Si se desacartan esos grupos o si la HP pareciera «desproporcionada» por su gravedad, realizar gammagrafía de ventilación/perfusión. Si muestra múltiples defectos de perfusión segmentaria, se plantea el diagnóstico del grupo 4: HPTC. El diagnóstico final de HPTC (y la valoración de idoneidad de una endarterectomía pulmonar) precisaráun angio-TAC, un CCD y una angiografía pulmonar selectiva. La TC también puede mostrar indicios que indiquen el grupo 1’: EVOP/HCP.
Si la gammagrafía de ventilación/perfusión es normal o únicamente revela defectos de perfusión dispersos, subsegmentarios, se hace el diagnóstico provisional del grupo 1: HAP o las enfermedades más raras del grupo 5. Pruebas diagnósticas específicas adicionales, que incluyen hematología, bioquímica, inmunología, serología y ultrasonografía, permitirán concretar el diagnóstico final. La Cuadro 11 describe el manejo adicional según la probabilidad de HAP, incluyendo indicaciones para el CCD.
La Cuadro 12 describe los grados de recomendación en la estrategia diagnóstica.
Evaluación de gravedad
Parámetros clínicos, ecocardiográficos y hemodinámicos
El pronóstico depende de la etiología de la HAP.
La clase funcional de la OMS (Cuadro 14) continúa siendo un poderoso indicador de supervivencia. En pacientes con HAPI o HAP heredable no tratados, los datos revelaron una supervivencia media de 6 meses para la CF IV de la OMS, 2,5 años para la CF III de la OMS y 6 años para las CF I y II de la OMS. Los extremos de edad (< 14 años o > 65 años), la capacidad de ejercicio reducida, el síncope, la hemoptisis y los indicios de insuficiencia cardiaca derecha también conllevan mal pronóstico en HAPI.
Los índices ecocardiográficos de mejor valor pronóstico según análisis multivariable son el derrame pericárdico, el área de la aurícula derecha, el índice de excentricidad del VI y el índice Doppler del VD.
Los valores hemodinámicos en reposo medidos durante el CCD predicen el pronóstico. Éstos incluyen la saturación del oxígeno de la AP, la presión auricular derecha, el GC, la RVP y una respuesta vasorreactiva marcada. La PAP es un elemento pronóstico, menos fiable puesto que puede disminuir hacia la fase final de la enfermedad cuando fracasa el VD.
Capacidad de ejercicio
Para valorar objetivamente la capacidad de ejercicio de pacientes con HAP se utilizan la prueba de marcha de 6 min (PM6M) y la prueba de ejercicio cardiopulmonar. Además de la distancia caminada, se registran la disnea durante el ejercicio (escala de Borg) y la saturación de O2 con pulsioxímetro digital. Distancias <
En la prueba de ejercicio cardiopulmonar se registran continuamente el intercambio gaseoso y la ventilación durante ejercicio. En la HAP, el consumo de O2 en el umbral anaeróbico y el pico de ejercicio se ven reducidos en relación con la gravedad de la enfermedad, así como la tasa pico de trabajo, el índice cardiaco pico, el pulso de O2 y la eficiencia ventilatoria. El análisis multivariable de los parámetros clínicos, hemodinámicos y de ejercicio mostraron que el consumo pico de O2 (< 10,4 ml O2/ kg/min) y la presión arterial sistólica pico (< 120 mmHg) durante el ejercicio predicen peor pronóstico en pacientes de HAPI de manera independiente.
Marcadores bioquímicos
Los marcadores bioquímicos son una herramienta no invasiva para la evaluación y observación de la disfunción del VD en pacientes con HP.
El ácido úrico sérico es un marcador del metabolismo oxidativo deficiente del tejido periférico isquémico. Altas concentraciones de ácido úrico se relacionan con menor supervivencia en pacientes con HAPI. Pero muchas veces a estos pacientes se les administra alopurinol y diuréticos lo que hace difícil interpretar esos valores. El péptido natriurético atrial y el péptido natriurético cerebral (BNP) inducen vasodilatación y natriuresis y son liberados por el miocardio en respuesta al aumento en la tensión de la pared. La insuficiencia del VD es la principal causa de muerte en la HAP y las concentraciones de BNP/NT-proBNP reflejan la gravedad de la disfunción del VD.
El valor medio basal del BNP (150 pg/ml) distingue entre los pacientes con buen o con mal pronóstico. El valor supramedio (> 180 pg/ml) se relaciona con un peor resultado a largo plazo. El BNP del plasma disminuye de manera significativa en los supervivientes, pero aumenta en los no supervivientes a pesar del tratamiento. En pacientes con HAP asociada a esclerodermia, el NT-proBNP menor de 553 pg/ml se relaciona con una mejor supervivencia.
Los aumentos en las concentraciones en plasma del NT-proBNP durante el seguimiento se han asociado con un peor pronóstico.
Las concentraciones plasmáticas del NT-proBNP se recomiendan para la estratificación del riesgo inicial y pueden tenerse en cuenta para supervisar los efectos del tratamiento, en vista de sus implicaciones pronósticas. Un NT-proBNP bajo y estable o que disminuye puede ser un marcador útil para un control eficaz de la enfermedad en la HAP.
Evaluación pronóstica completa. Ver Cuadro 14
Definición del estado del paciente
Según los resultados clínicos, invasivos y no invasivos El estado del paciente puede definirse como:
Estable y satisfactorio. Estos pacientes deben cumplir con la mayoría de los resultados de la columna «Mejor pronóstico» (Cuadro 15). No tienen signos clínicos de insuficiencia del VD, CF I o II de la OMS estable o sin síncope, marcha de 6 min >
Inestable y empeorando. Estos pacientes cumplen con lo enunciado en la columna «Peor pronóstico» (Cuadro 15). Hay evidencia de evolución de los síntomas y signos de insuficiencia del VD, empeoramiento de la CF de la OMS, sea de II a III o de III a IV, una marcha de 6 min <
pueden presentarse en este estado y contribuyen al deterioro clínico.
Objetivos del tratamiento y estrategia de seguimiento
Los objetivos del tratamiento se deben dirigir a cumplir con los criterios de la «Definición de estable y satisfactorio» y los resultados en la columna «Mejor pronóstico» de la Cuadro 15.
Los valores deberían ajustarse a cada paciente individualmente.
Por ejemplo, una PM6M >
Las recomendaciones para la evaluación de gravedad y seguimiento aparecen resumidas en la Cuadro 16.
Terapia
Medidas generales
Brindar consejo adecuado sobre las actividades generales de la vida diaria y necesitan adaptarse a la incertidumbre asociada a una enfermedad crónica grave que puede poner en peligro sus vidas.
• Actividad física y rehabilitación dirigida
Los pacientes deben permanecer activos dentro de los límites de sus síntomas. La dificultad leve para respirar es aceptable, pero deberían evitar esfuerzos que les produzcan gran dificultad para respirar, mareos o dolor torácico. Por ello, los pacientes deben evitar una actividad física excesiva que cause síntomas dolorosos, pero si se encuentran en baja forma física, deberían hacer ejercicios de rehabilitación dirigida. Hay evidencia creciente de la pérdida de masa muscular periférica en pacientes con HAP avanzada, y eso puede corregirse con un programa de rehabilitación definido.
• Embarazo, control de natalidad y terapia hormonal posmenopáusica
Existe documentación de que el embarazo está relacionado en el 30-50% de mortalidad en pacientes con HAP, por ello la HAP es una contraindicación para el embarazo. Los métodos anticonceptivos de barrera son seguros para la paciente, pero con un efecto impredecible. Los preparados sólo de progesterona, como el acetato de medroxiprogesterona y el etonogestrel, son efectivos para la anticoncepción y evitan consecuencias potenciales como aquellas incluidas en la minipíldora de la antigua generación.
Hay que recordar que el bosentán, antagonista de los receptores de la endotelina (ARE), puede reducir la eficacia de los agentes anticonceptivos orales.
El DIU de Mirena también es efectivo, raras veces produce una reacción vasovagal cuando es insertado, lo que puede ser mal tolerado en una HAP grave. También puede utilizarse una combinación de ambos métodos. La paciente que se embaraza debería ser informada del alto riesgo y se le debería hablar acerca de la interrupción del embarazo. Las pacientes que deciden continuar con su embarazo deberían recibir un tratamiento basado en terapias orientadas a la enfermedad, con un parto opcional deseado y con una colaboración estrecha efectiva entre los obstetras y el equipo a cargo de la HAP. No está claro si la aplicación de una terapia hormonal en las mujeres posmenopáusicas con HAP es aconsejable o no.
Puede tenerse en cuenta en casos de síntomas menopáusicos intolerables, además de anticoagulación oral.
• Desplazamientos
Los efectos fisiológicos conocidos de la hipoxia apuntan que durante el vuelo, debe administrarse O2 durante el vuelo debería considerarse para los pacientes con CF III o IV de la OMS y para aquellos con presión de O2 en sangre arterial habitualmente <8 kPa (60 mmHg). Un flujo de 2 l/min aumentará la presión de O2 inspirado hasta valores observados a nivel del mar. De igual manera, estos pacientes deberían evitar subir a altitudes superiores a los 1.500-
a 1600-2500m sobre el nivel del mar, por lo cual los pacientes con HP deberían utilizar oxígeno suplementario durante el vuelo si la duración es mayor a 2 horas. Hay que recomendar a los pacientes que viajen con información escrita acerca de su HAP y que sepan cómo contactar clínicas de HP locales en la proximidad del lugar al que viajan. • Apoyo psicosocial Muchos pacientes con HAP desarrollan ansiedad y depresión que les conducen a un deterioro de su calidad de vida. Hay que enviar al paciente a un psiquiatra o psicólogo en el momento
oportuno siempre que sea necesario. Los grupos de apoyo a los pacientes también pueden desempeñar un papel importante en este aspecto, y hay que aconsejar a los pacientes que se unan a ellos.
• Prevención de infecciones
Los pacientes con HAP son susceptibles de desarrollar neumonía, primera causa de muerte en el 7% de los casos.
Aunque no existen ensayos controlados, se recomienda vacunar a los pacientes contra la gripe y la neumonía neumocócica.
• Cirugía electiva
Se supone que la cirugía electiva tiene un mayor riesgo en pacientes con HAP. No está claro qué forma de anestesia es preferible, pero probablemente la epidural sea mejor tolerada que la anestesia general. Los pacientes que toman terapia oral pueden necesitar una conversión temporal a una i.v. o a un tratamiento nebulizador hasta que puedan tragar y absorber fármacos administrados por vía oral.
Las recomendaciones para las medidas generales están resumidas en la Cuadro 17.
Terapia de apoyo
Anticoagulantes orales
Dado que se encuentra una alta prevalencia de lesiones trombóticas vasculares en la autopsia de pacientes con HAPI y que se han reportado anomalías en las vías de coagulación y fibrinolíticas amén de la presencia de factores de riesgo no específicos para el tromboembolismo venoso (insuficiencia cardiaca e inmovilidad), se recomienda la anticoagulación oral en la HAP. La evidencia a favor de la anticoagulación oral generalmente es retrospectiva y se limita a pacientes con HAPI, HAP heredable y HAP causada por anorexígenos.
Los beneficios potenciales de la anticoagulación oral deberían contraponerse a los riesgos en los pacientes con otras formas de HAP, especialmente cuando hay mayor riesgo de hemorragia, como es el caso de la hipertensión portopulmonar con varices esofágicas graves.
El INR en HAPI debe mantenerse de
Diuréticos
La insuficiencia cardiaca derecha descompensada produce retención de líquidos, aumento en la presión venosa central, congestión hepática, ascitis y edema periférico. Aunque no hay ningún ECDA de diuréticos en la HAP, la experiencia clínica revela beneficios sintomáticos en pacientes con sobrecarga de fluidos tratados con esta terapia, 4970% de los pacientes con diagnóstico de HP son tratados con diuréticos. La elección y la dosis del diurético debe hacerla el médico de HAP. La adición de antagonistas de la aldosterona debería tenerse en cuenta. Es importante observar la función renal y la bioquímica sanguínea en los pacientes para evitar hipokalemia y los efectos de la disminución del volumen intravascular que llevan a la insuficiencia prerrenal.
Oxígeno
Hay datos que demuestran que la terapia con O2 nocturno no modifica la historia natural del síndrome de Eisenmenger avanzado.
La recomendación se basa en la evidencia de pacientes con EPOC; si la Pa O2 es consistentemente < 8 kPa (60 mmHg), se les aconseja usar O2 para llegar a un valor > 8 kPa durante al menos 15 h/día.
Debe considerarse la administración de O2 ambulatorio si hay evidencia de beneficio sintomático y de desaturación corregible durante el ejercicio. Desaturación nocturna (menor de 90%, en por lo menos 5% del total del registro nocturno de oximetría) se les recomienda la suplencia de oxígeno de la noche por 12 horas, siendo necesario evaluar un registro de oximetría que verifique la efectividad de la terapia.
A los pacientes con síntomas sugestivos de apnea e hipoapnea del sueño e HTP, por lo general leve o moderada, se les realiza una polisomnografía, en cuyo caso la terapia es con CPAP o BiPAP logrando reducciones de
Digoxina
La digoxina mejora el gasto cardiaco de forma aguda en la HAPI, pero se desconoce su eficacia cuando se administra de manera crónica.
Puede tomarse para enlentecer la respuesta ventricular en los pacientes con HAP que desarrollan taquiarritmias auriculares.
Terapia farmacológica específica
Bloqueadores de los canales de calcio
La hipertrofia, la hiperplasia y la vasoconstricción de las células de músculo liso contribuyen a la patogénesis de la HAPI, por ello se usan los vasodilatadores, principalmente los BCC. Se ha visto que sólo un pequeño número de pacientes con HAPI con respuesta favorable a la prueba aguda de vasorreactividad en el momento del CCD, mejoran con los BCC. Nunca se debe utilizar bloqueadores de canales de calcio sin realizar previamente pruebas de vasoreactividad pulmonar aguda con vasodilatadores.
El grado de reacción vasodilatadora no parece predecir una respuesta favorable a largo plazo a la terapia con BCC en pacientes con HAP asociada a ETC y estos pacientes a menudo no toleran bien una dosis alta de BCC.
Prostanoides
La prostaciclina, producida predominantemente por las células endoteliales, induce una potente vasodilatación de todos los lechos vasculares. Es el inhibidor endógeno más potente del agregado de plaquetas y también parece tener actividades citoprotectora y antiproliferativa.
En pacientes con HAP se ha demostrado la desregulación de las vías metabólicas de la prostaciclina , como se comprueba por la reducción de la expresión de la prostaciclina sintetasa en las arterias pulmonares y de los metabolitos urinarios de la prostaciclina.
El uso clínico de la prostaciclina en los pacientes con HAP se ha extendido, por la síntesis de análogos estables, que poseen diferentes propiedades farmacocinéticas, pero que comparten efectos farmacodinámicos similares desde el punto de vista cualitativo.
Septostomía atrial con balón (SAB)
Como los pacientes con síndrome de Eisenmenger y aquellos con HAPI y foramen oval persistente (FOP) tienen mejor supervivencia que los que no tienen FOP, se recomienda la SAB, como tratamiento para la HAPI. La apertura septal obliga al flujo de derecha a izquierda por la hipertensión derecha, lo que descomprime dichas cámaras, aumentando la precarga del VI y el gasto cardiaco.
Esto mejora tambien el transporte sistémico de O2 a pesar de la desaturación arterial de oxígeno y disminuye la hiperactividad simpática.
El procedimiento debe hacerlo un operador experimentado en el acceso endovascular e intracavitario y realizarse en una sala de hemodinamia debidamente equipada para ello. Se recomienda la técnica de septostomía más dilatación gradual con balón, y no la original de cuchilla. La SAB no está indicada en pacientes en fase terminal y que presenten una PAD basal media > 20 mmHg y una saturación de oxígeno en reposo < 80% en aire ambiente. Antes de plantearse la SAB los pacientes deben seguir una terapia médica óptima, que debe incluir un precondicionamiento con fármacos inotrópicos i.v. Existe clara evidencia de un beneficio en pacientes de CF IV de la OMS con insuficiencia cardiaca derecha refractaria a la terapia médica o con graves ataques sincopales. También se plantea en pacientes que esperan trasplante o cuando ya no hay terapia médica disponible. La HAPI grave ha sido la principal indicacioón de SAB en adultos, aunque otras incluyen HAP asociada a CC corregida quirúrgicamente, ETC, HPTC distal, EVOP y en la hemangiomatosis capilar pulmonar.
Trasplante
A pesar de una terapia específica, hasta el 25% de los pacientes con HAPI pueden no responder y el pronóstico de los que continúan en CF III o IV de la OMS, es malo, por ello el trasplante pulmonar es una opción importante. El pronóstico de la HAP varía según la etiología, y el de la HAP asociada a ETC es peor que el de la HAPI incluso cuando se trata con prostanoides, mientras que la supervivencia de los pacientes con HAP asociada a CC es mejor. El peor pronóstico es el de los pacientes con EVOP y hemangiomatosis capilar pulmonar debido a la falta de tratamientos médicos efectivos. Estos pacientes deberían ser incluidos en la lista para el trasplante una vez hecho el diagnóstico.
Los pacientes con un mal perfil pronóstico (Cuadro 15) a pesar de terapia médica máxima deberían ser remitidos a la lista de trasplante.
En HAP se ha realizado tanto el trasplante cardiopulmonar como el pulmonar doble, y cada centro ha desarrollado su propia escogencia
para cada paciente. Aunque la descarga del VD se reduce inmediatamente después de un trasplante pulmonar doble, la función sistólica del VD y la función diastólica del VI no mejoran de inmediato y la inestabilidad hemodinámica es un problema común en el primer periodo postoperatorio. Tanto el único como el doble se han llevado a cabo, aparentemente con una supervivencia similar. Sin embargo, cualquier complicación que ocurra en el aloinjerto posterior al trasplante pulmonar único está asociada a una hipoxemia grave. En la actualidad, la gran mayoría de los pacientes de todo el mundo reciben trasplantes pulmonares bilaterales, según lo demuestran las cifras del Registro de la Sociedad de Trasplantes de Corazón y Pulmón. Aunque los pacientes con síndrome de Eisenmenger causado por cortocircuitos simples han sido tratados con trasplante pulmonar aislado y reparación del defecto cardiaco, los pacientes con defectos septales ventriculares pueden obtener un mejor resultado con el trasplante cardiopulmonar. La supervivencia global a los 5 años después de un trasplante es del 40-45% para la HAP; con evidencia de una buena calidad de vida.
Algoritmo de tratamiento
En la figura 2 se muestra un algoritmo de tratamiento para pacientes con HAP. Las clases de recomendación y los niveles de evidencia para los tratamientos de HAP también se pueden consultar en la Cuadro 23. La definición de la respuesta clínica a los tratamientos aparece en la Cuadro 24.Las interacciones potenciales entre fármacos están en la Cuadro 22.
• El algoritmo de tratamiento es solo para pacientes del grupo
El planteamiento inicial propuesto tras el diagnóstico de HAP consiste en la adopción de las medidas generales, el comienzo de la terapia de apoyo y la referencia a un centro experto.
• La prueba de vasorreactividad aguda debe realizarse a todos los pacientes con HAP del grupo 1, aunque los pacientes con HAPI, HAP heredable y HAP asociada al uso de anorexígenos son los que más probabilidades tienen de manifestar una respuesta aguda positiva y de beneficiarse de la terapia con altas dosis de BCC. Los pacientes vasorreactivos, deben tratarse con dosis altas de BCC; la respuesta debe confirmarse tras 34 meses de tratamiento.
• Como no existen comparaciones frente a frente entre diferentes compuestos, se puede proponer un tratamiento de primera línea no basado en la evidencia. Dependiendo la elección del fármaco de una variedad de factores, que incluyen el estado de aprobación, la vía de administración, el perfil de efectos secundarios, las preferencias del paciente y la experiencia del médico.
Algunos expertos todavía utilizan epoprostenol i.v. en pacientes de la CF III de la OMS debido a sus resultados en cuanto a supervivencia.
Se recomienda el epoprostenol i.v. continuo como terapia de primera línea para pacientes con HAP en la CF IV de la OMS debido a la mejor supervivencia en este subconjunto. El treprostinil s.c. e i.v. también ha sido aprobado para el tratamiento de pacientes de la CF IV de la OMS en EEUU. Aunque no se han realizado ECDA con el iloprost administrado i.v.
• Aunque en EUA el ambrisentán, el bosentán y el sildenafilo están aprobados en los pacientes de la CF IV de la OMS, en el ECDA de estos agentes se incluyó sólo un pequeño número de esos pacientes.
Por lo tanto, la mayoría de los expertos consideran estos tratamientos como de segunda línea en los pacientes gravemente enfermos.
• La terapia de combinación inicial también debería utilizarse en los pacientes de la CF IV de la OMS.
En los centros expertos también se plantea la terapia de combinación triple.
Cuidados terminales y cuestiones éticas
El curso clínico de la HP consiste en el deterioro progresivo con intervalos de episodios de descompensación aguda. Es difícil prever cuándo morirán los pacientes a causa de una insuficiencia cardiaca.
Se ha demostrado que los médicos a cargo de estos pacientes tienden a ser demasiado optimistas en su pronóstico y a menudo malinterpretan los deseos de sus pacientes.
La comunicación abierta y confidencial con los pacientes permite tener una planificación avanzada y hablar acerca de sus miedos, preocupaciones y deseos, lo que es esencial para ofrecer un buen cuidado. Las oportunidades de hablar sobre el pronóstico deberían surgir en el momento del diagnóstico inicial. El reconocimiento de que la reanimación cardiopulmonar en la HAP grave tiene malos resultados puede inducirr una orden de «no resucitar». Esto puede aumentar lal posibilidad de que los pacientes se encuentren en su lugar de cuidado preferido al final de sus vidas.
Los pacientes que se acercan al final de su vida necesitan una valoración frecuente de todas sus necesidades por parte de un equipo multidisciplinario. Hay que prestarles atención para controlar los síntomas dolorosos y recetar los fármacos apropiados al mismo tiempo que se retira la medicación innecesaria. También es vital un apoyo psicológico, social y espiritual. Se debe consultar al especialista en cuidados paliativos acerca de los pacientes cuyas necesidades estén más allá de la experiencia del equipo de HP.
Subgrupos especificos de hipertensión arterial pulmonar
Algunas de las enfermedades incluidas en el grupo 1HAP, aunque presentan similitudes con la HAPI, tienen suficientes diferencias y necesitan comentarios específicos. Estas enfermedades comprenden las formas de HAP y HAPA (Cuadro 4) como CC, ETC, hipertensión portal e infección por el VIH. El reconocimiento de estas diferencias es crucial ya que pueden influir no sólo en el enfoque del diagnóstico, sino también en el manejo global de la HAP.
Hipertensión arterial pulmonar pediátrica
La HAP pediátrica es similar a la enfermedad del adulto, aunque en un niño los pulmones aún estén desarrollándose. Se desconoce la incidencia y prevalencia exactas de la HAP en niños. No se ha confirmado que el pronóstico sea peor en niños con una vida media estimada en 10 meses, en comparación con los 2,8 años en adultos. Todas las formas de HAP incluidas en la clasificación clásica (Cuadro 4) han sido descritas en niños, pero la mayoría de los pacientes se presentan pero la mayoría de los pacientes se presentan con HAP asociada a CC o a las formas idiopática y heredable. En cambio, la prevalencia de la HAP asociada a ETC, hipertensión portal, infección por el VIH y fármacos y toxinas es más baja. Cada vez son más los pacientes con enfermedad pulmonar crónica de la prematuridad.
La HAP persistente del neonato también está clasificada dentro de la HAP. Su historia natural, tratamiento y resultado son lo suficientemente
diferentes para justificar su exclusión de esta discusión. No se han identificado diferencias claras entre los mecanismos involucrados en el desarrollo de la HAP en niños y adultos.
Diagnóstico
Los niños a menudo se presentan más enfermos que los adultos. Los síntomas comunes son la disnea, fatiga y desarrollo insuficiente.
El síncope es más frecuente en los niños, pero la insuficiencia franca del VD es un acontecimiento tardío y el niño puede morir súbitamente antes de que ésta aparezca. Se aconseja realizar un estudio diagnóstico similar al que se hace en adultos. Incluso si algunas asociaciones son raras, deberían excluirse antes del diagnóstico definitivo. Es esencial obtener una historia familiar y personal minuciosa, con detalles del embarazo, el parto y del período posnatal. Puede obtenerse la PM6M y la prueba de ejercicio cardiopulmonar, pero se necesita experiencia y deben adaptarse a la edad. Para realizar el diagnóstico, se necesitan las pruebas del CCD y de vasorreactividad, hechas de la misma manera que en los adultos. En los niños estos procedimientos pueden requerir anestesia general, lo que aumenta los riesgos.
Terapia
La respuesta a la terapia es difícil de prever. El algoritmo terapéutico infantil es similar al utilizado en adultos, aunque falten ECDA específicos. Pocos estudios se han publicado para confirmar la dosis exacta de nuevas terapias aplicables en niños. La terapia debe incluir un seguimiento estricto.
El tratamiento rápido de cualquier infección de las vías respiratorias superiores o inferiores es esencial debido al peligro de un deterioro rápido. El uso de anticoagulación es controversial ya que no hay estudios disponibles para niños. El uso de la aspirina en vez de dicumarínicos también resulta polémico. El acuerdo consiste en anticoagular a los pacientes con insuficiencia cardiaca derecha clara.
Los BCC se utilizan en los respondedores, pero mantener un seguimiento estricto es obligatorio, ya que la terapia a largo plazo puede fracasar. Sí hay datos disponibles con bosentán, y su farmacocinética fue evaluada en un estudio. Recientemente la Agencia Europea del Medicamento aprobó una nueva formulación pediátrica.
Hasta ahora no hay datos disponibles sobre los antagonistas selectivos del receptor de la endotelina A.
El sildenafilo ha demostrado alguna eficacia, y un ECDA está en camino para definir la dosis y la eficacia.
Cada vez más pacientes pediátricos reciben una terapia de combinación, aunque todavía falta evidencia. La septostomía auricular y el cortocircuito de Pott son posibles en los niños, con buenos resultados. Al igual que en los adultos, la curación de la HAP únicamente se consigue con un trasplante pulmonar, pero la falta de donantes apropiados es un problema grave.
Hipertensión arterial pulmonar asociada a cortocircuito cardiaco congénito (grupo 1 de la clasificación clínica de la HP)
Si la RVP se aproxima o excede la resistencia vascular sistémica, el cortocircuito se invierte (síndrome de Eisenmenger).
Diagnóstico
De todos los pacientes con CC, aquellos con síndrome de Eisenmenger son los más gravemente afectados en términos de intolerancia al ejercicio. Es posible que la preservación de la función del VD resulte en mejor supervivencia, puesto que el VD no recibe remodelado al nacer y permanece hipertrofiado. El VD también se ve aliviado por el cortocircuito de derecha a izquierda, manteniendo el GC sistémico, a costa de la hipoxemia y la cianosis.
Los pacientes con CC (sobre todo aquellos sin cortocircuitos) también pueden desarrollar la HP causada por cardiopatía izquierda (grupo 2, Cuadro 4) o por enfermedades pulmonares concomitantes (grupo 3, Cuadro 4). En estos casos se recomienda un estudio diagnóstico completo, como se aconseja en el algoritmo de diagnóstico.
Terapia
La estrategia de tratamiento para los pacientes con HAP asociada a CC, y en particular para los sujetos con síndrome de Eisenmenger, se basa fundamentalmente en la experiencia clínica de los expertos.
Recientemente se ha propuesto un algoritmo de tratamiento específico similar al de la figura 2. Los pacientes con síndrome de Eisenmenger deberían tratarse en centros especializados. La educación del paciente, las modificaciones del hábito de vida y el conocimiento de factores de riesgo médicos potenciales son aspectos importantes de su manejo.
Los pacientes con síndrome de Eisenmenger pueden presentarse con deterioro clínico en diferentes circunstancias, que incluyen la cirugía no cardiaca con anestesia general, la deshidratación, las infecciones pulmonares y la gran altitud. Se recomienda evitar el ejercicio intenso, aunque parece que las actividades leves son beneficiosas. El embarazo está asociado a un alto riesgo tanto para la madre como para el feto, por lo que se desaconseja el embarazo y se recomienda la anticoncepción.
La terapia de O2 en casa a largo plazo, puede mejorar los síntomas, pero no se ha demostrado que modifique la supervivencia, al menos cuando se aplica sólo durante la noche. El uso de la terapia de O2 suplementario se recomienda en casos en los que genera un aumento consistente de la saturación arterial de oxígeno y reduzca los síntomas.
La eritrocitosis secundaria es beneficiosa para un transporte y entrega de O2 adecuados y debería evitarse la flebotomía sistemática. Si aparecen síntomas de hiperviscosidad, debe practicarse una flebotomía con sustitución isovolumétrica, normalmente cuando el hematocrito es > 65%. Habría que corregir la deficiencia de hierro. No hay datos claros que apoyen el uso de BCC en pacientes con síndrome de Eisenmenger, y el uso empírico de BCC es peligroso y debería evitarse.
Se ha demostrado que el bosentán mejora la PM6M y disminuye la RVP después de 16 semanas de tratamiento en pacientes con la CF III de la OMS.
Las experiencias anecdóticas con sildenafilo y tadalafilo, inhibidores de la fosfodiesterasa tipo 5, muestran resultados funcionales y hemodinámicos favorables en pacientes con HAP asociada a CC y síndrome de Eisenmenger. Se ha probado el uso de epoprostenol i.v. en pacientes con síndrome de Eisenmenger, mostrando efectos hemodinámicos favorables y en la capacidad de ejercicio, aunque las líneas centrales exponen a los pacientes a un riesgo de embolia paradójica y sepsis.
No hay datos disponibles con el uso de otros prostanoides.
Tampoco hay datos publicados acerca de la terapia de combinación, pero la base es la misma que en la HAPI.
El trasplante pulmonar o cardiopullmonar es una opción en casos especiales que no responden al tratamiento médico, pero está limitado por la disponibilidad de los órganos.
Las tasas de supervivencia a corto y largo plazo después del trasplante cardiopulmonar son similares a las de otras formas de HAP. La supervivencia prolongada estimada de los pacientes con síndrome de Eisenmenger hace difícil estimar la decisión de cuando se debería incluir a los pacientes en lista de espera.
Las recomendaciones para la hipertensión arterial pulmonar asociada a los cortocircuitos cardiacos congénitos están resumidas en la Cuadro 25.
Hipertensión arterial pulmonar asociada a enfermedad del tejido conectivo
La HAP asociada a ETC es la forma más común de HAP asociada y las más prevalente luego de HAPI; su principal exponente es la esclerosis sistémica, especialmente la variante cutáneo limitada (síndrome de CREST), pero también, lupus eritematoso sistémico, ETC mixta y, en menor grado, artritis reumatoide, dermatomiositis y síndrome de Sjögren.
La prevalencia exacta de ésta complicación es desconocida debido a diferentes criterios diagnósticos utilizados en los estudios, pero se estima entre 7 – 12% mediante la comprobación hemodinámica por CCD en grandes cohortes de pacientes con esclerosis sistémica.
En estos pacientes, la HAP puede ocurrir conjuntamente con fibrosis intersticial o como resultado de una arteriopatía pulmonar aislada. Además, la hipertensión venosa pulmonar por cardiopatía izquierda puede estar presente, por lo tanto, es fundamental determinar qué mecanismo impera, puesto que ello define el tratamiento.
Histológicamente, las lesiones presentadas por la vasculatura pulmonar, no difieren de las encontradas en HAPI, aunque hay una afectación más frecuente de las venas pulmonares. Los mecanismos fisiopatológicos que producen la HAP en pacientes con ETC se desconocen, sin embargo, la presencia de anticuerpos antinucleares, factor reumatoide, inmunoglobulina G y depósitos de fracción de complemento en las paredes de los vasos pulmonares sugieren la acción de un mecanismo inmunológico.
Diagnóstico
En comparación con la HAPI, los pacientes con ETC y HAP son principalmente mujeres (proporción mujeres/varones, 4:1), mayores (media de edad al diagnóstico, 66 años) y pueden presentar desórdenes concomitantes (fibrosis pulmonar, cardiopatía izquierda). Su supervivencia es más corta, de hecho, el riesgo de muerte no corregido para HAP asociada a la esclerosis sistémica en comparación con la HAPI es de 2,9 y los indicadores de resultados son los mismos que para la HAPI (PAD, PAP y IC). Los síntomas y la presentación clínica son muy similares a los de la HAPI, inclusive, pacientes aislados que parecen tener HAPI en verdad padecen ETC al realizar las pruebas inmunológicas.
La TC de alta resolución es útil para evaluar la presencia de enfermedad pulmonar intersticial asociada y dentro de las pruebas de función pulmonar, la reducción aislada de la capacidad de difusión de monóxido de carbono es una anomalía frecuente en la esclerosis sistémica asociada a la HAP.
La proporción de respondedores en la prueba vasodilatadora aguda es más baja que en la HAPI.
Terapia
El tratamiento de pacientes con HAP asociada a ETC es más complejo que el de los pacientes con HAPI. La terapia inmunosupresora que combina glucocorticoides y ciclofosfamida puede resultar en una mejoría clínica en pacientes con HAP asociada a lupus eritematoso sistémico o ETC mixta. La respuesta favorable a largo plazo al tratamiento con BCC en pacientes vasorreactivos es menor que en la HAPI. Debido al riesgo-beneficio de la anticoagulación oral no comprendido del todo en estos pacientes, su administración debe ser individualizada.
Hipertensión arterial pulmonar asociada a hipertensión portal
La HAP es una complicación muy conocida de las enfermedades hepáticas. La hipertensión portal, más que el desorden hepático, parece ser el principal factor de riesgo determinante para desarrollar una HAP. La enfermedad no es poco común, ya que la HAP asociada a la hipertensión portal (también llamada hipertensión portopulmonar) representa cerca del 10% de la población con HAP. Se cree que el 1-2% de los pacientes con enfermedad hepática e hipertensión portal desarrollan HAP, pero la prevalencia de la HAP puede alcanzar el 5% entre los pacientes con enfermedad hepática avanzada evaluados para trasplante de hígado. La patogenia no está clara y puede estar relacionada con sustancias tóxicas provenientes del tracto gastrointestinal no eliminadas por el hígado debido a los cortocircuitos portosistémicos, dañando así el endotelio pulmonar. Otra posiilidad es que un estado de alto GC genere la HAP.
Diagnóstico
El cuadro clínico de los pacientes con hipertensión portopulmonar puede ser indistinguible del de los pacientes con HAPI o puede incluir combinaciones de síntomas y signos de la enfermedad hepática subyacente.
La exploración ecocardiográfica para la detección de la HP en pacientes con enfermedades hepáticas es apropiada en pacientes sintomáticos y/o en candidatos a un trasplante de hígado. En todos los casos con una PAP sistólica aumentada debería realizarse un CCD para clarificar los cambios hemodinámicos subyacentes y definir las implicaciones pronósticas y terapéuticas. Los pacientes con hipertensión portopulmonar tienen un GC significativamente alto y unas RVP y resistencia vascular sistémica significativamente más bajas, en comparación con los pacientes con HAPI. En un estudio retrospectivo, los pacientes con hipertensión portopulmonar tuvieron una tasa de supervivencia mayor que los pacientes con HAPI, aunque sobre esto no hay acuerdo.
Terapia
La hipertensión portopulmonar es parte del espectro de enfermedad de la HAP y en general a estos pacientes hay que tratarlos de manera similar a aquellos con otras formas de HAP, al mismo tiempo que se tiene en cuenta la presencia de enfermedad hepática y sus consecuencias para su manejo. El algoritmo de tratamiento (Fig. 2) también puede aplicarse en este marco con adaptaciones.
La HAP puede aumentar sustancialmente el riesgo asociado al trasplante de hígado y normalmente la HAP es una contraindicación si la PAP media es ≥
Las recomendaciones para la hipertensión arterial pulmonar asociada a la hipertensión portal están resumidas en la Cuadro 26.
Hipertensión arterial pulmonar asociada a infección por el virus de la inmunodeficiencia humana
La HAP es una complicación infrecuente entre los pacientes con VIH, sin embargo, en comparación con individuos sanos su prevalencia es mayor, estimada en 0,1 – 0,5% con una incidencia estimada de 0,1% al año. A pesar que la terapia antirretroviral altamente efectiva y el manejo agresivo de las infecciones han aumentado la esperanza de vida en estos pacientes, la prevalencia e incidencia de la HAP ha permanecido similar al periodo previo a su uso, por lo que el impacto del tratamiento antirretroviral altamente efectivo sobre la HAP es discutido.
La patogénesis de la HAP relacionada con el VIH continúa siendo incierta y no se han descubierto partículas virales en las lesiones plexiformes complejas de estos pacientes lo que sugiere que puede existir una acción viral indirecta sobre factores de inflamación y el factor de crecimiento, que actúen como desencadenantes en un paciente predispuesto.
Diagnóstico
La HAP relacionada con el VIH tiene una presentación clínica similar a la HAPI y pueden presentar otros factores de riesgo para la HAP, tales como hepatopatías (hepatitis B o C), exposición a fármacos y toxinas o embolismo pulmonar debido a toxicomanía i.v. En el momento del diagnóstico, el 71-81% de los pacientes se encuentran en una CF de la OMS avanzada. Es más probable que los pacientes con HAP relacionada con el VIH sean varones y toxicómanos por vía i.v.
Más del 80% se encuentran bien controlados con la terapia antirretroviral altamente activa por lo que el recuento de CD4 no parece ser un factor de riesgo para su desarrollo.
Se recomienda realizar un ecocardiograma en pacientes con VIH y disnea inexplicable para detectar complicaciones cardiovasculares asociadas al VIH incluyendo HAP. La confirmación con un CCD es obligatoria para establecer el diagnóstico de la HAP relacionada con VIH y ausencia de cardiopatía izquierda.
La HAP es un factor de riesgo de muerte independiente en pacientes infectados por el VIH, con una sobrevida a 3 años del 21% en la mayor parte de los casos (CF III/IV de la OMS), en comparación con 84% en pacientes ligeramente sintomáticos.
Terapia
El tratamiento de la HAP relacionada con el VIH no está consolidado en comparación con otras formas de HAP debido a que solamente 3 ECDA (1 que utilizó prostanoide beraprost activo por vía oral y otros 2 con ambrisentán, antagonista de receptor de endotelina) permitieron la inclusión de pacientes con HAP relacionada con el VIH representando menos del 5% de la población total, sin embargo, se recomienda el mismo algoritmo de tratamiento que los pacientes con HAPI tomando en consideración las comorbilidades e interraciones farmacológicas.
La anticoagulación no está recomendada de forma sistemática debido a mayor riesgo de hemorragia, anticipando adherencia al tratamiento e interacción entre fármacos. Los pacientes con HAP relacionada con el VIH parecen no ser respondedores a las pruebas de vasorreactividad por lo que no deberían recibir BCC. Varios estudios no controlados indican que el epoprostenol i.v., el treprostinil s.c. y el iloprost inhalado pueden mejorar la tolerancia al ejercicio, la hemodinamia y los síntomas en la HAP relacionada al VIH.
En el caso del sildenafilo, la dosis debería ajustarse si el ritonavir y el saquinovir se coadministran, debido a la interacción entre ambos fármaco (Cuadro 22). Por lo general, se considera que la infección por el VIH es un criterio de exclusión para el trasplante pulmonar, incluso si algunos centros cuentan con un programa específico.
Enfermedad venooclusiva pulmonar y hemangiomatosis capilar pulmonar (grupo 1’)
Tanto la EVOP como la hemangiomatosis capilar pulmonar son enfermedades poco comunes, con menos de 200 casos reportados mundialmente, pero cada vez más se las reconoce como causantes de HAP. Se han incluido en un subgrupo específico de la clasificación clínica (Cuadro 4, grupo 1’) por las diferencias patológicas, clínicas y terapéuticas con respecto a otras formas de HAP incluidas en el grupo 1. Se ha informado de la ocurrencia familiar de la EVOP y se ha descubierto una mutación del receptor de proteínas morfogenéticas óseas tipo 2 en un paciente con esta enfermedad. Estos hallazgos hacen pensar que la HAP y la EVOP pueden representar diferentes manifestaciones fenotípicas de un único espectro de enfermedad. A diferencia de la HAPI, hay un predominio masculino en la EVOP y el pronóstico es peor.
Enfermedad venooclusiva pulmonar
Diagnóstico
El diagnóstico de la EVOP puede confirmarse con alta probabilidad combinando la sospecha clínica, el examen físico, la broncoscopia y los estudios radiológicos. Este enfoque no invasivo puede evitar la biopsia pulmonar (estándar para confirmar el diagnóstico de EVOP) en la mayoría de los casos. La principal sintomatología es disnea de esfuerzo y de fatiga, indistinguible de la HAPI. El reconocimiento físico puede revelar acropaquia digital y crépitos bibasales a la auscultación pulmonar, hechos inusuales en otras formas de HAP.
Una serie de casos indica que los pacientes con EVOP tienen hipoxemia más grave y una capacidad de difusión del monóxido de carbono mucho menor que aquellos con otras formas de HAP. Esto puede explicarse por la presencia de edema pulmonar intersticial crónico típico de la EVOP y/o un estado de bajo GC y/o la presencia de un foramen oval persistente.
La radiografía torácica puede revelar líneas B de Kerley e infiltrado intersticial periférico, además de otros signos típicos de la HP.
El examen a realizar es la tomografía axial de alta resolución. Los hallazgos típicos que indican una EVOP son la presencia de líneas septales subpleurales más engrosadas, opacidades centrolobulares en vidrio esmerilado (que contrastan con la distribución panlobular hallada en la HAPI) y linfadenopatía mediastínal. La asociación de estos 3 hallazgos resultó ser el 100% precisa para la EVOP en casos de HAP, con una sensibilidad del 66%. Además, su presencia se correlaciona cercanamente con el riesgo de edema pulmonar a causa de la terapia con epoprostenol.
Dado que la EVOP puede estar asociada a una hemorragia alveolar oculta por lo que la broncoscopia con lavado broncoalveolar puede ser una herramienta útil en la estrategia diagnóstica. La presentación hemodinámica de la EVOP es parecida a la de la HAPI. En gran medida, la PEP es casi invariablemente normal porque los cambios patológicos tienen lugar en pequeñas vénulas que no afectan a las venas pulmonares más grandes. La prueba de vasorreactividad puede complicarse por el edema pulmonar agudo.
Terapia
No hay ninguna terapia médica establecida para la EVOP. Lo que es más importante, los vasodilatadores y, sobre todo, los prostanoides deben utilizarse con gran precaución debido al alto riesgo de edema pulmonar. No existen informes de mejoría clínica constante en pacientes individuales tratados con estos fármacos. Tampoco hay datos en relación con el uso de terapias médicas más nuevas, como los ARE o los inhibidores de la fosfodiesterasa tipo 5 en el tratamiento de la EVOP y la hemangiomatosis capilar pulmonar. Por lo
tanto, se recomienda referir con prontitud una vez establecido su diagnóstico a un centro de transplante pulmonar para su evaluación así como para la administración de tratamiento específico por personal con amplia experiencia en donde se informe a los pacientes de sus riesgos.
La septostomía atrial puede considerarse, pero normalmente se ve limitada por la hipoxemia. La única terapia curativa para la EVOP y la hemangiomatosis capilar pulmonar es el trasplante pulmonar, al igual que sucede con la HAPI, no hay informes sobre la recurrencia de la enfermedad después del trasplante.
Hemangiomatosis capilar pulmonar
Enfermedad muy rara que puede ser difícil de diferenciar de la EVOP. Los aspectos de diagnóstico y tratamiento son muy similares.
A menudo, sólo el reconocimiento patológico permite distinguir las 2 enfermedades.
Hipertensión pulmonar por cardiopatía izquierda
(grupo 2)
La HP conlleva un pronóstico malo para los pacientes con insuficiencia cardiaca crónica. En un estudio, la tasa de mortalidad después de 28 meses de seguimiento fue del 57% en pacientes con HP moderada en comparación con el 17% en pacientes sin HP. Además, los pacientes que tienen una RVP mayor de 6-8 unidades Wood (480-640 dinas·s·cm–5 ) tienen también un mayor riesgo de sufrir insuficiencia postoperatoria del VD tras un trasplante de corazón.
Diagnóstico
El enfoque diagnóstico de la HP causada por cardiopatía izquierda es similar al de la HAP; la ecocardiografía Doppler es la mejor herramienta de exploración. Debería sospecharse la disfunción diastólica del VI en presencia de aurícula izquierda dilatada, fibrilación auricular, cambios característicos en el perfil del flujo mitral, perfil del flujo venoso pulmonar, señales Doppler del anillo mitral e hipertrofia del VI. Datos de la evaluación del doppler tisular muestran que la proporción E/E’ de la velocidad de flujo precoz de la válvula mitral (E) dividida por la velocidad de prolongación diastólica temprana (E’) está íntimamente relacionada con las presiones de llenado del VI: cuando la proporción E/E’ es superior a 15, las presiones de llenado del VI se elevan y cuando la proporción es inferior a 8, las presiones de llenado del VI son bajas; si 15 > E/E’ > 8, se precisan investigaciones no invasivas adicionales.
Las características clínicas y ecocardiográficas típicas de la HP asociada a disfunción diastólica del VI son:
• Manifestaciones clínicas: mayores de 65 años, aumento de la presión arterial sistólica, aumento de la presión de pulso, obesidad, síndrome metabólico, hipertensión, enfermedad arterial coronaria, diabetes mellitus, fibrilación atrial
• Ecocardiograma: crecimiento atrio izquierdo, remodelación concéntrica del VI (pared engrosada > 0,45), hipertrofia del VI, hallazgos que indiquen aumento de la presión de llenado del VI
• Evaluación posterior al ecocardiograma: respuesta de síntomas con diuréticos, aumento exagerado de la presión arterial con el ejercicio, re-evaluación de la rediografía de tórax consistente con insuficiencia cardíaca.
Aunque un aumento en las presiones de llenado del lado izquierdo pueden calcularse con la ecocardiografía Doppler, las mediciones invasivas de la PEP o la presión diastólica final del VI pueden ser necesarias para confirmar el diagnóstico de HP causada por cardiopatía izquierda (véase cateterismo cardiaco). La PEP y la presión diastólica final del VI pueden ser «seudonormales», sobre todo cuando los pacientes han sido tratados con diuréticos. En este marco, cambios hemodinámicos del volumen en ejercicio se han propuesto para identificar disfunción del VI, pero estas herramientas diagnósticas precisan una mayor estandarización.
La HP conlleva un pronóstico malo para los pacientes con insuficiencia cardiaca crónica. En un estudio, la tasa de mortalidad después de 28 meses de seguimiento fue del 57% en pacientes con HP moderada en comparación con el 17% en pacientes sin HP. Además, los pacientes que tienen una RVP mayor de 6-8 unidades Wood (480- 640 dinas·s·cm–5 ) tienen también un mayor riesgo de sufrir insuficiencia postoperatoria del VD tras un trasplante de corazón.
Diagnóstico
El enfoque diagnóstico de la HP causada por cardiopatía izquierda es similar al de la HAP; la ecocardiografía Doppler es la mejor herramienta de exploración. Debería sospecharse la disfunción diastólica del VI en presencia de aurícula izquierda dilatada, fibrilación auricular, cambios característicos en el perfil del flujo mitral, perfil del flujo venoso pulmonar, señales Doppler del anillo mitral e hipertrofia del VI. Datos de la evaluación del doppler tisular muestran que la proporción E/E’ de la velocidad de flujo precoz de la válvula mitral (E) dividida por la velocidad de prolongación diastólica temprana (E’) está íntimamente relacionada con las presiones de llenado del VI: cuando la proporción E/E’ es superior a 15, las presiones de llenado del VI se elevan y cuando la proporción es inferior a 8, las presiones de llenado del VI son bajas; si 15 > E/E’ > 8, se precisan investigaciones no invasivas adicionales.
Las características clínicas y ecocardiográficas típicas de la HP asociada a disfunción diastólica del VI son:
• Manifestaciones clínicas: mayores de 65 años, aumento de la presión arterial sistólica, aumento de la presión de pulso, obesidad, síndrome metabólico, hipertensión, enfermedad arterial coronaria, diabetes mellitus, fibrilación atrial
• Ecocardiograma: crecimiento atrio izquierdo, remodelación concéntrica del VI (pared engrosada > 0,45), hipertrofia del VI, hallazgos que indiquen aumento de la presión de llenado del VI
• Evaluación posterior al ecocardiograma: respuesta de síntomas con diuréticos, aumento exagerado de la presión arterial con el ejercicio, re-evaluación de la rediografía de tórax consistente con insuficiencia cardíaca.
Aunque un aumento en las presiones de llenado del lado izquierdo pueden calcularse con la ecocardiografía Doppler, las mediciones invasivas de la PEP o la presión diastólica final del VI pueden ser necesarias para confirmar el diagnóstico de HP causada por cardiopatía izquierda (véase cateterismo cardiaco). La PEP y la presión diastólica final del VI pueden ser «seudonormales», sobre todo cuando los pacientes han sido tratados con diuréticos. En este marco, cambios hemodinámicos del volumen en ejercicio se han propuesto para identificar disfunción del VI, pero estas herramientas diagnósticas precisan una mayor estandarización.
Un gradiente transpulmonar elevado (PAP media menos PEP media) > 12 mmHg indica cambios intrínsecos en la circulación pulmonar anulando el aumento pasivo en la PEP. En algunos pacientes, puede resultar difícil distinguir la HAP de la HP asociada a disfunción del VI, sobre todo en aquellos con valores de la PEP en el límite (15-18 mmHg).
Un gradiente transpulmonar elevado (PAP media menos PEP media) > 12 mmHg indica cambios intrínsecos en la circulación pulmonar anulando el aumento pasivo en la PEP. En algunos pacientes, puede resultar difícil distinguir la HAP de la HP asociada a disfunción del VI, sobre todo en aquellos con valores de la PEP en el límite (15-18 mmHg).
No se ha demostrado la utilidad de las concentraciones plasmáticas del BNP para el diagnóstico de la cardiopatía izquierda en presencia de HP, porque ambas enfermedades fisiopatológicas pueden elevarlo. Tampoco la utilidad de la evaluación hemodinámica con esfuerzo o sobrecarga de fluidos. El papel, la importancia y el marco de la prueba farmacológica permanecen poco claros en la HP causada por cardiopatía izquierda, aunque se recomienda identificar a los pacientes con mayor riesgo de insuficiencia postoperatoria aguda del VD.
Terapia
En la actualidad no existe ninguna terapia específica para la HP causada por cardiopatía izquierda. Existen algunos fármacos (diuréticos,
La historia de la terapia médica para la insuficiencia cardiaca está llena de ejemplos en los que los fármacos tuvieron efectos positivos
en los resultados, pero al final resultaron ser perjudiciales, como los inhibidores de la fosfodiesterasa tipo 3.
Por lo tanto no se recomienda el uso de fármacos específicos para la HAP hasta que estudios a largo plazo no ofrezcan datos seguros, en particular de la HP «desproporcionada» asociada a cardiopatía izquierda (Cuadro 3). La reducción constante de la HP se espera en semanas o meses en la mayoría de los pacientes eficazmente operados de la enfermedad de la válvula mitral, aunque la HP representara un factor de riesgo para la cirugía.
Hipertensión pulmonar por enfermedades
Pulmonares y/o hipoxemia (grupo 3)
Incluye una gama de entidades que mediante la vasoconstricción hipóxica, estrés mecánico de la hiperinsuflación pulmonar, pérdida de capilares, inflamación, efectos tóxicos del cigarro o su combinación, pueden desarrollar HP. Histológicamente pueden presentar hipertrofia de la media, proliferación obstructiva de la íntima de las arterias pulmonares distales y varios grados de destrucción del lecho vascular en enfisema, así como, zonas de fibrosis. En la EPOC, la presencia de la HP está asociada a supervivencia corta y a episodios frecuentes de exacerbación. La incidencia de HP significativa en estos pacientes con antecedente de una hospitalización por exacerbación de su insuficiencia respiratoria es 20% y aumenta considerablemente en EPOC avanzado (> 50%). La HP es un factor pronóstico malo en las enfermedades pulmonares intersticiales, con una prevalencia estimada de 32 – 39% y la PAP es el indicador de mortalidad más importante.
Diagnóstico
Los indicaciones para ecocardiografía de tamizaje en busca de HP en la EPOC y en enfermedades pulmonares intersticiales incluyen:
a) exclusión de HP significativa; b) evaluación de cardiopatía izquierda concomitante, y c) selección de pacientes para el CCD.
El diagnóstico definitivo de la HP depende de las mediciones que se obtengan en el CCD. Sus indicaciones en enfermedad pulmonar avanzada son: a) diagnóstico de HP en candidatos a tratamientos quirúrgicos (trasplante, reducción del volumen pulmonar); b) posible HP «desproporcionada » potencialmente susceptible de entrar a formar parte de un ECDA con terapia específica de fármacos para la HAP; c) episodios frecuentes de insuficiencia del VD, y d) estudio ecocardiográfico no concluyente en casos con un alto grado de sospecha.
Terapia
En la actualidad no hay ninguna terapia específica para la HP asociada a la EPOC o a las enfermedades pulmonares intersticiales.
Se ha demostrado que la administración de O2 a largo plazo reduce parcialmente la evolución de la HP en la EPOC. No obstante la PAP nunca regresa a sus valores normales con este tratamiento y las anomalías estructurales de los vasos pulmonares permanecen inalteradas.
No está claro el papel que tiene el tratamiento de O2 a largo plazo en la evolución de la HP en las enfermedades pulmonares intersticiales.
No se recomienda aplicar el tratamiento con vasodilatadores convencionales porque pueden afectar al intercambio de gas debido a la inhibición de la vasoconstricción pulmonar hipóxica y a su falta de eficacia tras un uso a largo plazo. La experiencia publicada
con terapia específica de fármacos para la HAP es escasa y consiste en la valoración de los efectos agudos y en pequeñas series de estudios no controlados. El tratamiento de elección para pacientes hipoxémicos con EPOC o enfermedades pulmonares intersticiales y HP asociada es la terapia de O2 . Los pacientes con HP «desproporcionada » causada por enfermedades pulmonares (caracterizada por disnea insuficientemente explicada por trastornos mecánicos pulmonares y por una PAP media ≥ 40-45 mmHg en reposo) deberían acudir a centros expertos y participar en ensayos clínicos orientados hacia la terapia específica de fármaco para la HAP.
En la actualidad no se aconseja el uso de una terapia centrada en la HAP en pacientes con EPOC o enfermedades pulmonares intersticiales y una PAPmedia < 40 mmHg porque no hay datos sistémicos con respecto a su seguridad o eficacia. y estos pacientes deberían tratarse dentro de los ensayos clínicos siempre que fuera posible. En la actualidad no se ha aprobado ninguna terapia médica ni en Europa ni en los Estados Unidos para la HPTC. El trasplante pulmonar bilateral es una opción para los casos avanzados que no encajan en la EAP.
convencionales porque pueden afectar al intercambio de gas debido a la inhibición de la vasoconstricción pulmonar hipóxica y a su falta de eficacia tras un uso a largo plazo. La experiencia publicada con terapia específica de fármacos para la HAP es escasa y consiste en la valoración de los efectos agudos y en pequeñas series de estudios no controlados. El tratamiento de elección para pacientes hipoxémicos con EPOC o enfermedades pulmonares intersticiales y HP asociada es la terapia de O2 . Los pacientes con HP «desproporcionada » causada por enfermedades pulmonares (caracterizada por disnea insuficientemente explicada por trastornos mecánicos pulmonares y por una PAP media ≥ 40-45 mmHg en reposo) deberían acudir a centros expertos y participar en ensayos clínicos orientados hacia la terapia específica de fármaco para la HAP.
En la actualidad no se aconseja el uso de una terapia centrada en la HAP en pacientes con EPOC o enfermedades pulmonares intersticiales y una PAPmedia < 40 mmHg porque no hay datos sistémicos con respecto a su seguridad o eficacia y estos pacientes deberían tratarse dentro de los ensayos clínicos siempre que fuera posible. En la actualidad no se ha aprobado ninguna terapia médica ni en Europa ni en los Estados Unidos para la HPTC. El trasplante pulmonar bilateral es una opción para los casos avanzados que no encajan en la EAP.
Hipertensión pulmonar tromboembólica crónica (grupo 4)
Ciertas enfermedades se asocian a un aumento del riesgo de HPTC, incluyendo esplenectomía previa, la presencia de un cortocircuito ventriculo-auricular para el tratamiento de la hidrocefalia, desórdenes mieloproliferativos y enfermedades inflamatorias intestinales crónicas.
Diagnóstico
A los pacientes con embolia pulmonar aguda que muestren signos de HP o disfunción del VD en cualquier momento durante su estancia hospitalaria se les debe practicar un ecocardiograma de seguimiento después del egreso (normalmente, 3-6 meses) para determinar si la HP se ha solucionado o no.
En pacientes con HP inexplicada se recomienda realizar una gammagrafía pulmonar de ventilación/ perfusión para excluir la HPTC. Una gammagrafía de ventilación/ perfusión normal descarta la HPTC.
Para determinar la estrategia terapéutica adecuada, normalmente se utilizan herramientas invasivas como el CCD y la angiografía pulmonar tradicional.
La angiografía coronaria es aconsejable en candidatos a tromboendarterectomía y con factores de riesgo de enfermedad coronaria.
Con el fin de minimizar los riesgos y los procedimientos repetidos, estas investigaciones tendrían que ser llevadas a cabo en un centro experto en vez de en los hospitales de referencia. El diagnóstico final de la HPTC se basa en la presencia de HP precapilar (PAP media ≥ 25 mmHg, PEP ≤ 15 mmHg, RVP > 2 unidades Wood) en pacientes con múltiples trombos/émbolos oclusivos crónicos/ organizados en las arterias pulmonares elásticas (principal, lobular, segmentaria, subsegmentaria).
Terapia
Los pacientes con HPTC deberían recibir anticoagulación por vida, normalmente con antagonistas de vitamina K regulados a un RNI entre 2,0 y 3,0.
Los trombos organizados proximales representan la indicación ideal, mientras que las obstrucciones más distales pueden impedir un procedimiento eficaz. Tras una intervencion efectiva, la RVP puede descender drásticamente y la hemodinámica pulmonar pueden casi normalizarse. Un centro tiene suficiente experiencia en este campo si realiza al menos 20 operaciones EAP al año con una mortalidad < 10%.
La terapia específica de fármacos para la HAP puede desempeñar un papel en los pacientes de HPTC, principalmente para 3 casos diferentes: a) si el paciente no es un candidato para la cirugía; b) si se considera que el tratamiento preoperatorio es apropiado para mejorar las hemodinámicas, y c) si el paciente se presenta con HP sintomática residual/ recurrente tras la endarterectomía pulmonar.
Varios estudios clínicos no controlados parecen indicar que los prostanoides, los ARE y los inhibidores de la fosfodiesterasa tipo 5 pueden producir beneficios hemodinámicos y clínicos en los pacientes con HPTC, sin importar si se consideraba a esos pacientes operables o inoperables.
El único ensayo clínico aleatorizado y controlado con placebo que ha abordado hasta ahora la seguridad y la eficacia del tratamiento médico fue el estudio BENEFIT, que investigó los efectos del bosentán en pacientes con HPTC inoperable durante un periodo de 16 semanas. Este estudio reveló un descenso significativo en la RVP en el grupo del bosentán, pero no se produjo ningún cambio en la PM6M, en la clase funcional ni en el tiempo de empeoramiento clínico.
El trasplante pulmonar bilateral es una opción para los casos avanzados que no encajan en la EAP.
Bibliografía
1. Guía de práctica clínica para el diagnóstico y tratamiento de la hipertensión pulmonar. Rev Esp Cardiol, 2009; 62: 1464 e. Disponible en versión electrónica: www.revesp.cardiol.org. [ Links ]
2. Guía para el diagnóstico y tratamiento de la Hipertensión Arterial Pulmonar. 2010. Asociación Colombiana de Neumología y Cirugía de Tórax. [ Links ]
3. Jamieson SW, Kapelanski DP, Sakakibara N, et al. Pulmonary endarterectomy: experience and lessons learned in 1,500 cases Ann
4. Favaloro R, Peradejordi M.A., Gómez C.,
5. Oudiz R.J. Pulmonary hippertension associated with left - sided heart diseane. Clin Chest Med 2007; 28: 233-41. [ Links ]
6. Reichenberger F., Voswinckel R., Encke B, Rutsch M, Fechtalli EE., Schmehl et al. Long-term treatment with sildenafil in chronic thrombo-embolic pulmonary hippertension. Eur Respir J 2007; 30: 922-927. [ Links ]
Recibido 09-IV-2012. Aceptado 11-IX-2012


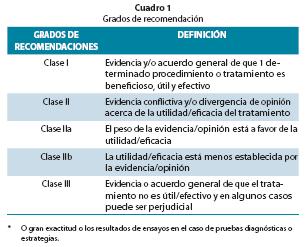{kind=link}
{kind=link}
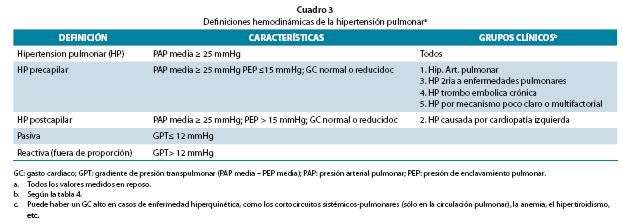{kind=link}
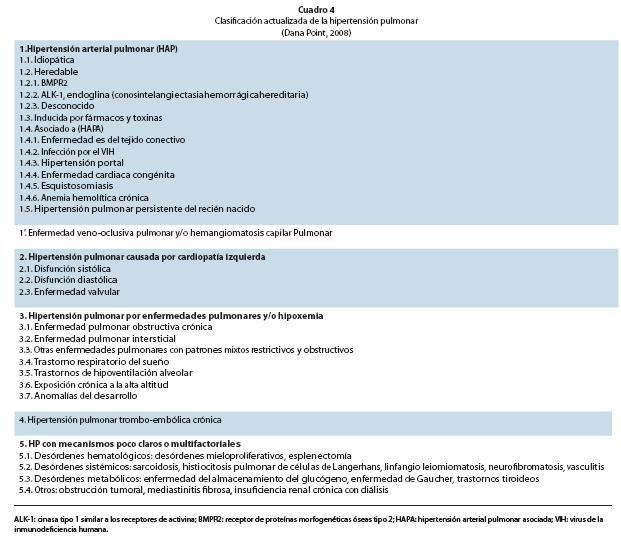{kind=link}
{kind=link}
{kind=link}
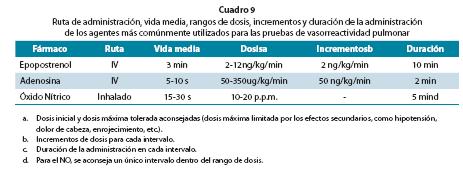{kind=link}
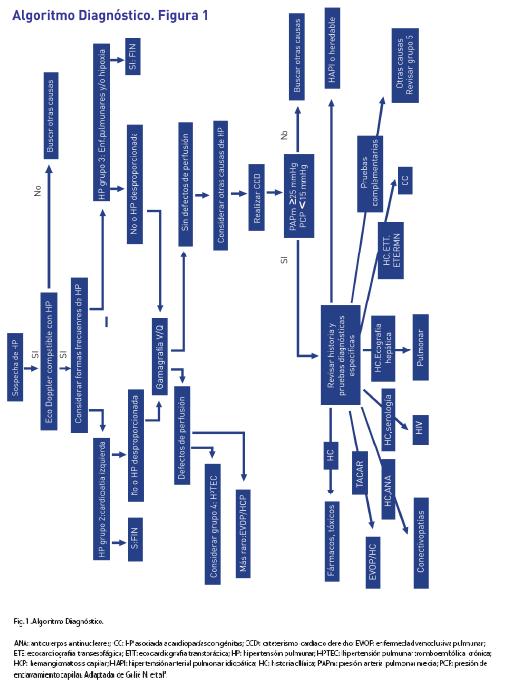{kind=link}
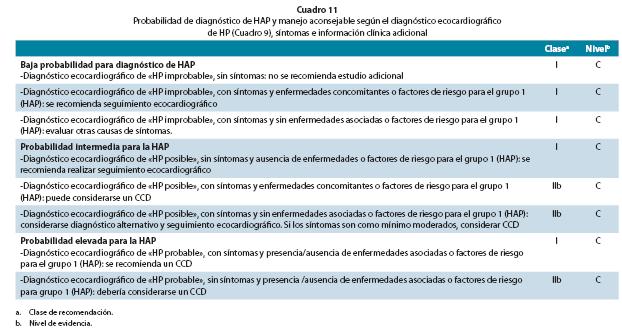{kind=link}
{kind=link}
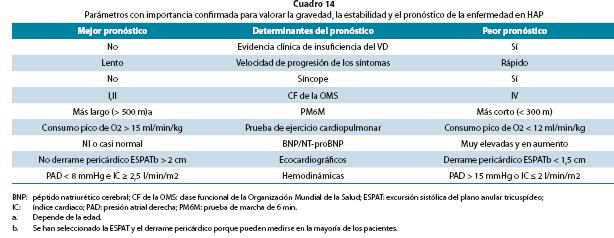{kind=link}
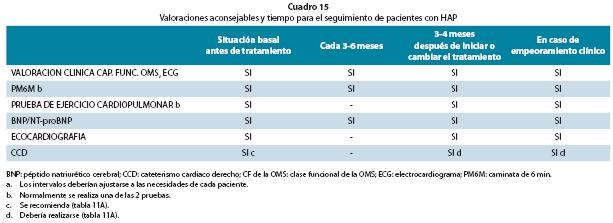{kind=link}
{kind=link}