











 (pdf)
(pdf)




 SciELO
SciELO  SciELO
SciELO 
 Permalink
PermalinkRevista Costarricense de Cardiología
ISSN 1409-4142
Taquicardia ventricular sostenida en un paciente con distrofia muscular miotónica
Vladimir Putvinskya, Oswaldo Gutiérrezb
a. Médico residente en medicina interna, Hospital Rafael A. Calderón Guardia, San José, Costa Rica.
b. Servicio de Cardiología, Hospital México, San José, Costa Rica.
Resumen
Se presenta el caso de un paciente con distrofia miotónica o enfermedad de Steinert, quien presentó taquicardia ventricular sostenida con daño miocárdico y que requirió terapia con cardiodesfibrilador implantable.
Palabras clave: Distrofia miotónica –Taquicardia ventricular – desfibrilador implantable.
Abstract
The case of a male patient with myotonic dystrophy or Steinert disease, with myocardial involvement, who developed sustained ventricular tachycardia, is presented herein. He underwent implantation of an automatic defibrillator.
Key words: Myotonic Dystrophy – Ventricular Tachycardia – Implantable defibrillator.
Abreviaturas: DMM: distrofia muscular miotónica; CTG: citosina – timidina – guanina; EKG: electrocardiograma; TV: taquicardia ventricular; FV: fibrilación ventricular.
Introducción
La enfermedad de Steinert o distrofia muscular miotónica (DMM) es la distrofia muscular más frecuente en adultos. Se presenta en 1 de 8000 nacimientos y tiene una prevalencia en población adulta mundial de 2.1-14.3/100 000 personas. Se divide en 3 formas: congénita, clásica y mínima. La forma clásica es la más común y los síntomas se manifiestan entre la segunda y la cuarta décadas de la vida con una progresión lenta a través del tiempo. Es una enfermedad heredofamiliar autosómico-dominante;la mutación consiste en una expansión inestable de la secuencia repetitiva (hasta 2000 veces) del trinucleotido CTG en el gen de DMM-kinasa del cromosoma 19q13.3.
Además del compromiso muscular, la enfermedad afecta los ojos (cataratas), sistema endocrino (diabetes, trastornos tiroideos, hipogonadismo), sistema nervioso central (retardo mental, trastornos cognitivos), sistema gastrointestinal (disfagia, constipación, colelitiasis, pseudoobstrucción) y el corazón1,2.
El compromiso cardiaco es frecuente y predominan alteraciones del sistema de conducción His-Purkinje. Todavía no está claro el mecanismo biomolecular de los trastornos cardiacos. El compromiso miocárdico es raro y se manifiesta por alteraciones de función ventricular diastólica o sistólica. En material de autopsia, histológicamente, se observa fibrosis, infiltración grasa y atrofia del sistema de conducción. En la mitad de los pacientes se reportan arritmias supraventriculares y ventriculares. Se han descrito bradicardia sinusal, bloqueos atrioventriculares, complejos prematuros atriales y ventriculares, fibrilación y flutter atrial, taquicardia y fibrilación ventricular. En el último decenio los mecanismos de muerte súbita y su prevención en los pacientes con DMM han sido temas de particular interés1,2,6.
Caso clínico
Masculino de 54 años, portador de DMM diagnosticada a los 35 años, con antecedentes familiares de esta enfermedad (3 hermanos y 3 hermanas). El paciente refirió haber tenido hace 2 años "arritmia ventricular", probablemente complejos ventriculares prematuros, que fue tratada con aspirina 100 mg/d, enalapril 20 mg/d y amiodarona 200 mg/d. Hace un mes antes se le diagnosticó una catarata del ojo izquierdo.
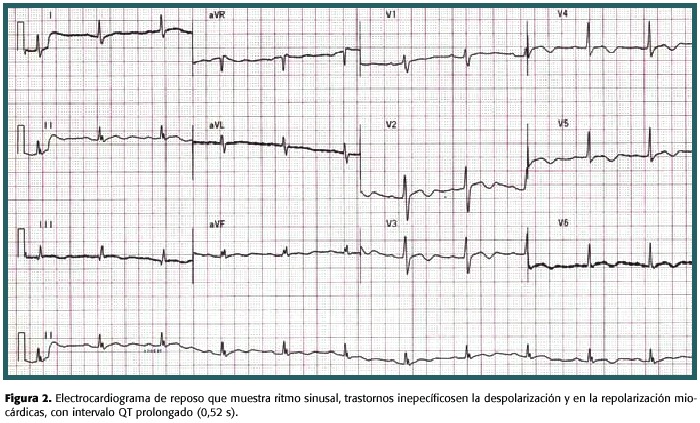
Consultó al servicio de emergencias del Hospital Calderón Guardia por un cuadro de palpitaciones, dolor torácico anginoso de media hora de evolución, asociado a críodiaforesis y lipotimia. La presión arterial estaba en 90/60 mmHg y la frecuencia cardiaca en 180 lat/min. El electrocardiograma (EKG) de ingreso mostró una taquicardia ventricular monomorfa que resolvió espontáneamente varios minutos después (fig. 1). El EKG durante ritmo sinusal mostró trastornos inespecíficos en la repolarización ventricular, intervalo QT prolongado pero sin evidencia de isquemia miocárdica. La radiografía de tórax no mostró cardiomegalia, ni otras alteraciones. El manejo y los estudios en el servicio de emergencias se enfocaron hacia un síndrome coronario agudo. No se detectó elevación de las enzimas cardiacas y el hemograma, la creatinina, el nitrógeno ureico, los electrolitos y las pruebas de función tiroidea se reportaron dentro de límites normales.
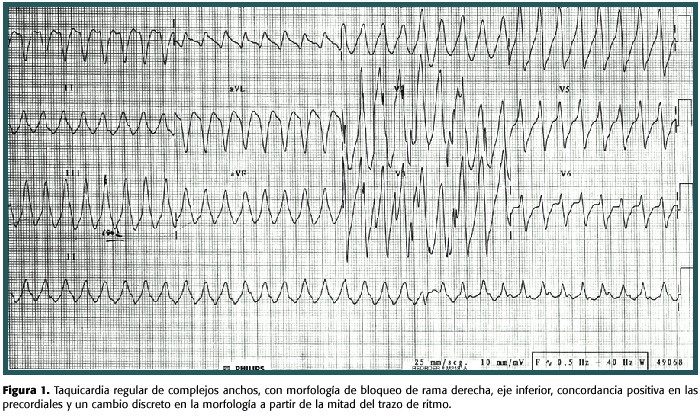
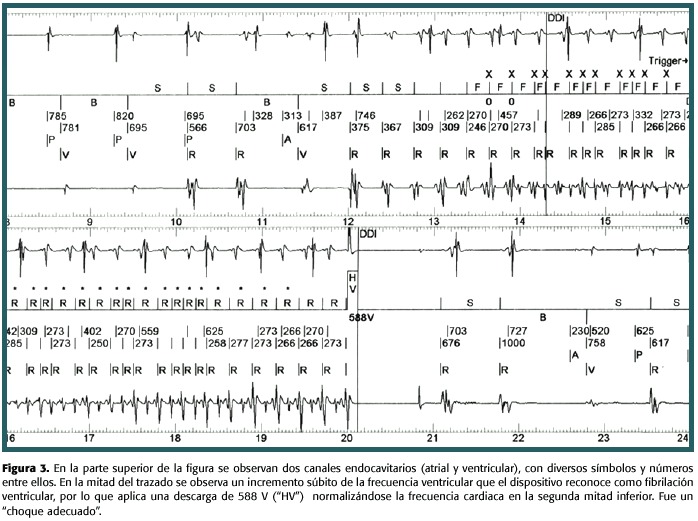
Se le administraron 900 mg amiodarona en infusión intravenosa durante 24 horas y luego 200 mg bid PO. El paciente evolucionó sin síntomas durante su estancia hospitalaria, sin presentar nuevos episodios de palpitaciones, ni dolor torácico. En el estudio Holter de 24 horas se observaron 638 complejos ventriculares prematuros de 14 formas y 4 dupletas. Los hallazgos ecocardiográficos fueron hipokinesia difusa leve, grosor del ventrículo izquierdo limítrofe, disfunción diastólica tipo 1 y fracción de eyección 45-50%. La arteriografía coronaria no demostró lesiones obstructivas y fue egresado asintomático. Posteriormente se le implantó un cardiodesfibrilador y suspendió la amiodarona y aproximadamente un año después, se presentó en el servicio de urgencias por múltiples terapias del dispositivo en el lapso de unas 12 horas. Una vez interrogado este mediante telemetría, se observó que habían ocurrido múltiples episodios de taquicardia ventricular sostenida, los cuales fueron hemodinámicamente bien tolerados y adecuadamente tratados (fig. 3).Se incrementó el número de terapias de "estimulación antitaquicardia" y se volvió a indicar amiodarona (200 mg/d PO), evolucionando sin eventos en los siguientes 2 meses.
Discusión
En los pacientes con DMM es frecuente observar trastornos del sistema de conducción y específicamente del sistema His-Purkinje. En pacientes asintomáticos, en estadíos tempranos de la enfermedad pueden observarse defectos de conducción menores y su progresión a defectos más severos de conducción puede debutar con disnea, lipotimia, síncope y muerte súbita.
En diferentes estudios se reporta la prevalencia de PR prolongado se reporta en 20-40% y de complejos QRS anchos en 5-25%3. En un estudio se reportó que la presencia de "potenciales tardíos" traduce prolongación del intervalo HV más que variaciones en los potenciales ventriculares4.
Las indicaciones para implantación de marcapasos son las usuales, pero en algunos centros se indican en forma temprana, por ejemplo, si se encuentra un intervalo HV prolongado (>70 ms) en el estudio electrofisiológico, con alto valor predictivo5.
Las taquiarritmias son frecuentes en pacientes con DMM y se detectan con el EKG de reposo o con registro Holter de 24 horas. Hasta en un 25% de los pacientes puede haber flúter o fibrilación atrial. En los estudios de mortalidad de los pacientes con DMM, la causa cardiovascular está en el segundo lugar (alrededor del 30%) después de las enfermedades respiratorias. Se considera que las causas de muerte súbita son asistolia, taquicardia ventricular (TV) fibrilación ventricular (FV) y disociación electromecánica6.
La TV monomorfa, la TV polimorfa y la FV están bien documentadas en estos pacientes, tal como en el presente caso. En los pacientes con un cuadro clínico sugestivo de TV previa, con o sin historia familiar de muerte súbita, debe considerarse realizar un estudio electrofisiológico. Puede inducirse TV en el 18% de los pacientes, incluso en ausencia de arritmias ventriculares en el registro Holter. La TV inducible más frecuente es la polimórfica no sostenida, pero también se ha reportado la TV polimórfica sostenida, TV monomórfica sostenida y no sostenida y FV7.
La terapia antiarrítmica varía. En los casos en los que se documente "TV por reentrada entre ramas de haz de His" la ablación por catéter es el tratamiento de elección7,8. El desfibrilador implantable se utiliza según las recomendaciones del ACC/AHA/NASPE1,2,9 pero no está definida su indicación en los pacientes asintomáticos con TV inducible en el estudio electrofisiológico.
Hay muy poca información acerca del uso de antiarrítmicos como sotalol o amiodarona y su eficacia comparativa. Se está levando a cabo un estudio prospectivo y multicéntrico en Italia (el estudio RAMYD) para evaluar el riesgo de arritmias y complicaciones cardiológicas en pacientes con DMM10.
En conclusión, la DMM es una enfermedad que puede producir compromiso cardiaco con arritmias fatales y el médico tratante tiene que sospecharlo y detectarlo ya que potencialmente puede ser mortal, como en el presente caso.
Referencias
1. G. Pelargonio, A Dello Russo, et al. Myotonic dystrophy and the heart. Heart 2002; 88: 665–670. [ Links ]
2. Braunwalds Heart Disease: A Textbook of Cardiovascular Medicine, 7th ed. Saunders. Philadelphia. 2005. Págs. 2146-2151. [ Links ]
3. Oloffson B, Forsberg H, Andersson S, et al.Electrocardiographic findings in myotonic dystrophy. Br Heart J 1988; 59: 47-52. [ Links ]
4. Babuty D, Fauchier L, Tena-Carbi D, et al. Significance of late potentials in myotonic dystrophy. Am J Cardiol 1999; 84:1099-101. [ Links ]
5. Lazarus A, Varin J, Duboc D. Final results of the French diagnostic pacemaker study in myotonic dystrophy. PACE 2002; 25: 599. [ Links ]
6. Mathieu J, Allard P, Potvin L, et al. A 10 year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 1999; 52: 1658- 62. [ Links ]
7. Merino JL, Carmona JR, Fernandez-Lozano I, et al. Mechanisms of sustained ventricular tachycardia in myotonic dystrophy. Circulation 1998; 98: 541-6. [ Links ]
8. Ramírez CJ, Rodríguez DA, et al. Myotonic Dystrophy and Bundle-Branch Re-Entrant Tachycardia. Rev Esp Cardiol 2002; 55: 1093-1097. [ Links ]
9. Gregoratos G, Abrams J, Epstein AE, Freedman RA, Hayes DL, Hlatky MA, Kerber RE, Naccarelli GV, Schoenfeld MH, Silka MJ, Winters SL. ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/NASPE Committee on Pacemaker Implantation). 2002. Available at: www.acc.org/clinical/guidelines/pacemaker/pacemaker.pdf. Consultado el 12-03-2007 [ Links ]
10. RAMYD Study - Evaluation of Arrhythmic Risk in Myotonic Dystrophy, Study protocol available at: www.clinicaltrials.gov (febrero de 2007). Consultado el 12-03-2007. [ Links ]
a. Dirección para correspondencia: Servicio de Medicina Interna, Hospital Rafael Calderón Guardia, Bo. Amón, San José, Costa Rica. Teléfono (506)825-3586. Fax (506)231-3856. Ap. Postal 471-1300
Correo electrónico: vlputvinsky@hotmail.com

