










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Cardiología
Print version ISSN 1409-4142
Rev. costarric. cardiol vol.8 n.1 San José Jan. 2006
El calcio en los miocitos cardíacos y su papel en las miocardiopatías.
Guido Ulate Monteroa, Adriana Ulate Camposb
Resumen
El papel que desempeña el calcio en los miocitos cardíacos abarca un gran número de funciones, desde su rol en el acoplamiento excitación-ccontracción hasta su papel de segundo mensajero en las diversas vías de señalización, algunas de las cuales se activan en procesos que afectan la integridad del tejido miocárdico y que tienen que ver con el crecimiento y la apoptosis de los miocitos y que al final, son los que determinan la evolución de la mayoría de las cardiomiopatías.
En esta revisión se abordan los mecanismos fisiológicos en las células de músculo cardíaco en los que este ión juega un papel determinante y los cambios que se presentan en ciertas cardiopatías como la miocardiopatía arritmogénica del ventrículo derecho, las miocardiopatías asociadas a alteraciones en el receptor de rianodina y la miocardiopatía dilatada. Se revisa también la farmacodinamia de los agentes inotrópicos positivos que actúan sensibilizando los miofilamentos del sarcómero al calcio.
Palabras clave: Calcio, miocitos cardíacos, miocardiopatías, levosimendán, enfermedad cardíaca.
Abstract
The rol played by calcium in cardiomyocytes includes a great number of functions like the excitation-contraction coupling as well as a second messenger in diverse signaling pathways. Some of these pathways are activated in processes that affect the cardiac tissue integrity and also participate in cell growth and apoptosis, determining the poor prognosis that characterize the majority of the cardiomyopathies.
In this review,the mechanisms inside the cardiomyocytes in which participates calcium and the pathophysiological changes observed in some cardiac diseases like arrhythmogenic right ventricular cardiomyopathy, catecholaminergic polymorphic ventricular tachycardia, idiopathic dilated cardiomyopathy, and others, are described. The action mechanism of positive inotropic drugs that act as sarcomere myofilaments sensitizers is also reviewed.
Key words: Calcium, cardiomyocytes, cardiomyopathies, levosimendan, heart disease.
Introducción
A lo largo de la evolución, el calcio (Ca2+ ) dentro de las células se ha convertido en un importante mensajero que realiza gran cantidad de funciones fisiológicas fundamentales como son el acople excitación-ccontracción en las fibras musculares, la comunicación entre las células, la secreción, el crecimiento e inclusive la muerte1.
Evolutivamente, el papel primordial del calcio se ha desarrollado gracias a la aparición de una serie de proteínas del interior de la célula, muchas de ellas con actividad enzimática, que son sensibles a los cambios en la concentración intracelular de este ión. Sin embargo, además de la aparición de esas proteínas, también ha sido necesario el desarrollo de canales iónicos en las membranas celulares que permitan el ingreso del Ca2+ de acuerdo con su gradiente electroquímico, así como la presencia de almacenes que favorecen, ante determinadas circunstancias, su liberación y con ello el aumento de su concentración citosólica. Este incremento permite que el calcio interaccione con las proteínas encargadas de llevar a cabo diferentes funciones fisiológicas celulares1-3.
En la presente revisión se explican los diferentes procesos fisiológicos que ocurren en los miocitos, en los cuales el calcio juega un papel decisivo4,5, así como las consecuencias clínicas que conlleva la alteración de tales procesos. Para tal efecto, se revisará inicialmente la estructura de los miocitos y los pasos involucrados en el mecanismo del acople excitación-contracción.
Aspectos estructurales de los miocitos cardíacos
La función primordial del corazón es el bombeo eficiente de sangre gracias a los ciclos de contracción-relajación, que de forma coordinada, ejecutan los miocitos.
Los miocitos cardíacos poseen un núcleo grande y oval situado en la parte central6,7 y se caracterizan por la presencia de uniones terminoterminales altamente especializadas, que se denominan discos intercalares.Los discos intercalares tienen porciones transversales, en las que abundan fascias adherentes, desmosomas, así como uniones comunicantes o "gap junctions". Estas últimas permiten el acople eléctrico así como el paso de pequeñas moléculas (<11 kDa) e iones8.
La membrana de los miocitos se denomina sarcolema y el citoplasma se llama sarcoplasma. El sarcolema presenta invaginaciones hacia el sarcoplasma, que reciben el nombre de túbulos T o túbulos transversos. Los miocitos contienen las organelas típicas, destacándose la presencia de un sistema de retículo endoplasmático liso muy desarrollado que recibe el nombre de retículo sarcoplasmático (RS) y en su interior se acumula gran cantidad de calcio unido a proteínas llamadas calsecuestrina y calreticulina. Las estructuras formadas por el túbulo T y la cisterna que está a su lado se llaman díadas.
Una de la principales características de estas células es la presencia de bandas, algunas oscuras y otras claras, que se repiten continuamente a lo largo de la miofibrilla y cuya unidad funcional se conoce con el nombre de sarcómero9.
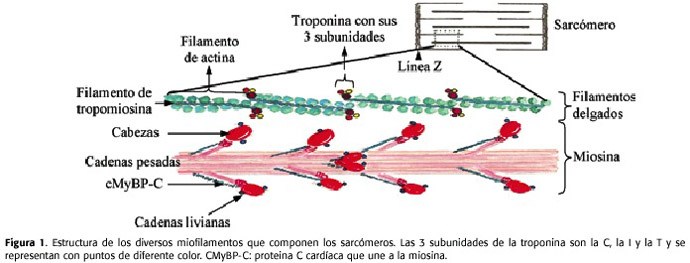
Un sarcómero está compuesto por diferentes tipos de filamentos (Figura 1)10:
1. Filamentos gruesos: compuestos por miosina. La miosina es una proteína formada por 6 cadenas polipeptídicas: 2 cadenas ligeras esenciales, 2 cadenas ligeras reguladoras y 2 cadenas pesadas, cuyos dominios amino-terminales forman una estructura globular, llamada cabeza de la miosina, donde se van a unir las 4 cadenas ligeras.
2. Filamentos delgados: compuestos por 3 tipos de proteínas: la actina, que forma una doble hélice a partir de la actina G; la tropomiosina que también tiene forma de hebra y se asocia a cada uno de los monómeros de la actina y la troponina, que está constituida por tres subunidades distintas: la troponina T (TnT) que se une a la molécula de tropomiosina; la troponina I (TnT) que está unida a la actina, en una posición que bloquea los centros de unión que existen en la actina para la miosina y la troponina C (TnT), la cual tiene dos dominios: uno de ellos, sería el correspondiente a la terminal amino y el otro a la terminal carboxilo. En cada uno de los dominios existen dos centros de unión al Ca 2+.
3. Filamentos intermedios como la titina, la desmina y la vimentina. La titina es una proteína fibrosa, una de las más largas que se conoce. Actúa como un muelle y tiene una secuencia de aminoácidos que permite que se produzcan las contracciones y se relaja en cuanto finaliza la contracción muscular.
Acople excitación-ccontracción en el músculo cardíaco
La secuencia de eventos (Figura 2) que se produce durante una contracción del músculo cardíaco es la siguiente: inicialmente, un potencial de acción se desplaza por el sarcolema, incluidos los túbulos T. Durante la fase de la meseta del potencial, se produce el ingreso de Ca2+ en los miocitos a través de los canales tipo L4 . Este Ca2+ que ingresa funciona como mensajero para producir la liberación de calcio del RS. El fenómeno se conoce como "liberación de Ca2+ inducida por Ca2+ " y ocurre a través de canales que están presentes en la membrana del RS y que se conocen con el nombre de receptores de rianodina tipo 2 (R y R2). Los R y R2 se localizan próximos a los canales de Ca2+ tipo L, formando unidades funcionales llamadas "couplon ", las cuales constan de aproximadamente 100 R y R2 junto con 25 canales tipo L. El Ca2+ que ingresa desde el exterior celular interactúa con los R y R2 y produce su apertura.
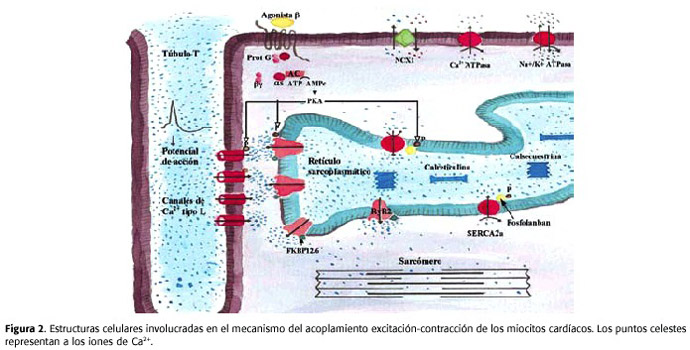
Al abrirse los R y R2, el calcio que se encuentra almacenado dentro del RS sale, con lo que aumenta la concentración de Ca2+ en el citosol. El Ca2+ liberado se une a la TnC. El complejo Ca2+ -troponina produce el desplazamiento de la tropomiosina del surco de la actina en que estaba ubicada. Ese desplazamiento deja descubiertos los sitios de la actina a los cuales se unen las cabezas de la miosina (puentes cruzados).
La interacción entre la actina y las cabezas de miosina permite que ocurra el ciclo de los puentes cruzados y de esa manera se produzca el acortamiento, es decir, la contracción. Para que se dé este ciclo, es necesario que se hidrolice el ATP y que las cabezas de la miosina interaccionen con los sitios descubiertos de la actina. La frecuencia de los ciclos de los puentes cruzados determina la velocidad de acortamiento del músculo.
Cuando cesan los potenciales de acción que recorren el sarcolema,la concentración de Ca2+ citosólico comienza a disminuir,provocando que la tropomiosina cubra nuevamente los sitios de la actina que interaccionan con los puentes cruzados. Al cubrirse los sitios de interacción, cesa el deslizamiento y el sarcómero recupera la longitud que tenía antes de la contracción, es decir, ocurre la relajación muscular. La concentración de Ca2+ citosólico disminuye por la recaptación de calcio en el RS debida a la Ca2+ -ATPasa presente en la membrana del RS de los miocitos, o por la salida de este catión de la célula, gracias a la Ca2+ -ATPasa del sarcolema y al antiportador 3Na+ /1Ca2+ (NCX1) también presente en el sarcolema. La actividad de la Ca2+ -ATPasa es regulada por una proteína llamada fosfolamban: cuando el fosfolamban está defosforilado, inhibe a esta bomba4,5,11-14.
Patología cardíaca asociada a alteraciones en el manejo intracelular del calcio
En los últimos años se han realizado nuevas investigaciones y se ha podido determinar de manera más clara, el papel fisiopatologico que juega el manejo inadecuado del calcio intracelular.Este conocimiento ha permitido una mejor comprensión sobre la presentación, la evolución y la respuesta a los medicamentos de estas enfermedades.
Sin embargo,es importante referirse a un punto que actualmente es motivo de gran investigación y controversia: ¿porqué si la concentración intracelular de calcio está constantemente aumentando de forma cíclica durante la sístole, no se desencadenan una serie de otros procesos intracelulares, que se han asociado a manejo inadecuado del calcio?. Muy recientemente, esta pregunta ha sido abordada por Wu15 y por Molkentin5 quienes postulan que el calcio tendría un papel en la contracción (global) y otro en la señalización intracelular (local). Dentro de las posibles explicaciones está la existencia de dos almacenes de Ca2+ diferentes: uno, presente en el RS, movilizado durante la contracción y el otro, aunque no está completamente definido, podría estar localizado en la membrana nuclear15. Aquí, la liberación de Ca2+ estaría determinada por receptores para el inositol trifosfato (IP3 ) presentes en la membrana nuclear, los cuales serían estimulados por aquellos mensajeros químicos que se unen a receptores en el sarcolema (GGPCRs) de los miocitos cardíacos y que terminan aumentando la producción y liberación de IP3.
El calcio liberado por efecto del IP3, formaría microambientes locales en los que se activa una cascada de eventos que incluyen a la kinasa II dependiente de calmodulina (CaMKII) y la salida del núcleo de la 5-deacetilasa de la histona (HDAC5). Esta última proteina normalmente actúa reprimiendo la activación transcripcional y favoreciendo la condensación del ADN. Cuando la HDAC5 es fosforilada por la CaMKII, sale del núcleo y con ello se activa la expresión de varios genes que determinan un programa de hipertrofia de los miocitos16. De esta manera, los miocitos podrían distinguir simultáneamente las señales globales de las locales, es decir las señales involucradas normalmente en el acople excitación-contracción de las señales que modulan la expresión genética.
Muy probablemente, serían estas últimas las que al final determinan la evolución de las miocardiopatías. En la tabla 1 se resumen algunas condiciones patológicas asociadas a poteínas relacionadas con el Ca2+ en el miocito.

Miocardiopatía arritmogénica del ventrículo derecho (MAVD).
Inicialmente, esta patología fue descrita como una enfermedad familiar del ventrículo derecho, caracterizada por arritmias ventriculares17,11 y la aparición progresiva de tejido fibroadiposo que reemplaza a los miocitos18. En la actualidad también se ha descrito en el ventrículo izquierdo19.
En el 50% de los casos existe historia familiar positiva20, comúnmente heredada de manera autosómica-ddominante con expresión variable y penetrancia incompleta20,21 y generalmente se diagnostica entre la segunda y quinta década de la vida. También existe una forma autosómica recesiva que se ha descrito asociada a la enfermedad de Naxos22 en la cual, además de la MAVD, se presentan problemas en la piel y en el pelo. La manera de presentación, aunque variable, suele incluir palpitaciones, síncope o muerte súbita23,24.
Los principales genes asociados a la etiología de este padecimiento son los que codifican para las siguientes proteinas de los miocitos: la placoglobina (JUP en 17q21), la desmoplaquina (DSM en 6p24), la placofilina-2 (PKP2 en 12p11) y el receptor de rianodina (RyR2 en 1q42). Las 3 primeras, son proteinas que se encuentran en los desmosomas de los discos intercalados, la última, es el canal a través del cual sale el Ca2+ del retículo sarcoplasmático durante la contracción.
Los desmosomas, como ya se dijo, son uniones especializadas que existen entre los miocitos cardíacos y que sirven para acoplar los elementos del citoesqueleto, como los filamentos intermedios, con el sarcolema. Su función es permitir la transmisión de la fuerza a toda la masa muscular, con lo cual se asegura la integridad mecánica del tejido cardíaco. En los desmosomas interaccionan 3 tipos de proteinas25: las cadherinas, las armadillo y las plaquinas (Figura 3). Las primeras son proteinas transmembranales, los dominios extracelulares interactúan con las cadherinas de la célula vecina permitiendo la adhesión; para que la adhesión funcione adecuadamente es necesaria la presencia de calcio. Las plaquinas son las proteinas que sujetan a los filamentos intermedios del citoesqueleto y las proteinas armadillo se ubican entre las cadherinas y las plaquinas.
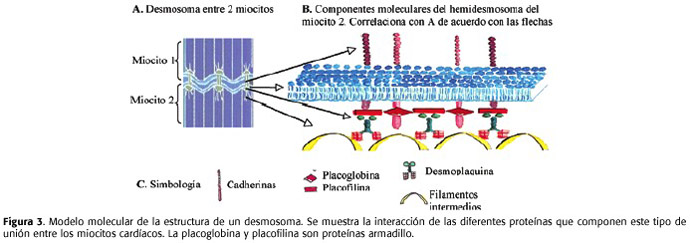
En la MAVD, las alteraciones estructurales debidas al defecto que presentan algunas de las proteinas que normalmente forman los desmosomas (como la JUP, la DSM y la PKP2) impiden la adecuada interacción entre miocitos y el mantenimiento de la integridad celular, lo cual termina induciendo el fenómeno de apoptosis26,27. Luego, los miocitos que mueren son reemplazados por tejido fibroadiposo19,28,29. Como ya se dijo, existe otra forma de MAVD en la cual la proteína afectada no forma parte de los desmosomas sino que se trata más bien del RyR2. Esta variante será tratada en el próximo apartado.
Miocardiopatías asociadas a alteraciones en el receptor de rianodina (RyR2)
Existen 3 entidades que se han asociado con un funcionamiento alterado de este receptor: la insuficiencia cardíaca, la taquicardia ventricular catecolaminérgica polimórfica (TVCP) y una variante de la MAVD, específicamente la tipo 2. En todas estas entidades, se presenta una pérdida constante ("goteo")de Ca2+ desde el RS, lo que lleva a la producción de posdespolarizaciones tardías que pueden conducir a la taquicardia ventricular12,14,31. La pregunta aquí es ¿por qué ocurre ese goteo desde el RS? Para responderla, se debe revisar con mayor detalle las características del RyR2 y su modulación.
El canal RyR2 es un tetrámero,cada subunidad pesa aproximadamente 550 kDa. Es el canal iónico más grande conocido hasta ahora. Contiene gran cantidad de sitios a los que se unen diversos ligandos que modulan su comportamiento, como por ejemplo el Ca2+, el Mg2+, el lactato, el ATP, la rianodina, la cafeína, los aminoglicósidos y el verapamil12. El Ca2+ es de importancia particular pues juega un papel primario en su activación durante el acople excitación-contracción. Por otro lado, el RyR2 se une a otras moléculas proteicas y conjuntamente forman un complejo de mayor tamaño14,32. Algunas de estas son la calmodulina (CaM)33,34, la calstabina-2 (FKBP12.635-37) y la calsecuestrina 32. La primera inhibe la actividad del RyR2 14 , la segunda produce su estabilización 12,32,37 y la tercera se relaciona con la capacidad de almacenar Ca2+ del RS14.
Inicialmente, Marks y cols38 demostraron que cuando el RyR2 era fosforilado por la proteína quinasa A (PKA), la calstabina-2 (FKBP12.6) se separaba del macrocomplejo, lo que aumentaba la probabilidad de apertura del canal de RyR2, la salida de Ca2+ y un incremento en la concentración citosólica de Ca2+ que podía ser responsable de la generación de arritmias. Posteriormente, estos hallazgos fueron confirmados.Por ejemplo,en la insufciencia cardíaca (Figura 2), la activación crónica del sistema simpático conduce a hiperfosforilación del RyR2, por ende separación de la FKBP12.6 y al goteo de Ca2+ desde el RS. Esto lleva a una depauperación del contenido de este ión dentro del RS, a una menor salida de Ca2+ durante la sístole, y por ende, a una pobre contractilidad del miocardio11,14.
La TVCP es una enfermedad descrita hace casi tres décadas39. Típicamente, los pacientes que la padecen desarrollan una taquicardia ventricular bidireccional o polimórfica que conduce a episodios de síncope o muerte súbita durante la liberación aumentada de catecolaminas desencadenada por emoción o ejercicio físico. En la mayoría de los casos se hereda de manera autosómica-dominante y el problema se ha rastreado hasta mutaciones en el gen del canal RyR2. Aproximadamente el 30% de los pacientes tiene historia familiar de muerte súbita juvenil o de síncope relacionado con el estrés40. Una variante de la enfermedad se hereda de manera autosómica recesiva y corresponde a una mutación en el gen de la calsecuestrina.
En estos pacientes con mutaciones en los canales RyR2, todo parece indicar40 que bajo condiciones basales o de reposo, el comportamiento de estos canales no es diferente al de los normales pero que, ante la activación del sistema simpático, su fosforilación por la PKA disminuye su afinidad por la proteína FKBP12.6 y como consecuencia se produce salida de Ca2+ del RS, aún en diástole.
Por último, la aparición de fármacos que estabilizan el canal RyR2, como el derivado de la 1,4-benzotiacepina (JTV519),se podrían convertir en una forma novedosa de tratar aquellos pacientes que presentan arritmias ventriculares originadas por defectos genéticos en este canal. De hecho, el beneficio clínico de los agentes betabloqueadores en el tratamiento de la insuficiencia cardíaca crónica tiene su explicación en la disminución de la fosforilación de los RyR241 que producen esos fármacos.
Miocardiopatía dilatada
Se caracteriza por el aumento de los volúmenes ventriculares y la disminución de la contractilidad sistólica, en ausencia de enfermedad coronaria42,43,44. Patológicamente, se puede observar leve hipertrofia de los miocitos, degeneración de ellos y fibrosis intersticial45. Con respecto a su etiología, se han involucrado diversos factores ambientales como las infecciones virales y la toxicidad por drogas, sin embargo, en una tercera parte de los casos existe un factor hereditario42,43. La transmisión genética es principalmente autosómica dominante42. En esta revisión abarcaremos los debidos a alteraciones genéticas.
La mayoría de los genes involucrados en la etiología de esta patología codifican para componentes del citoesqueleto45, para componentes del aparato contráctil46 como la miosina, la titina, la actina, la tropomiosina (Tm) la troponina T (TnT), la troponina C (TnC) y la proteina C cardíaca que une la miosina (cMyBP-C) y para componentes del mecanismo "liberación de calcio inducida por calcio"47,48 como el fosfolamban y la SERCA2a. Las mutaciones en los genes que codifican para los filamentos del sarcómero pueden causar fenotipos muy contrastantes, lo que ha sido utilizado en el análisis de los posibles mecanismos fisiológicos y de las relaciones entre estructura y función49. Por ejemplo, tanto la cardiomiopatía hipertrófica como la dilatada pueden producirse por mutaciones en el gen que codifica para la cMyBP-C50. La cMyBP-C (Figura 1) es una proteina accesoria de los sarcómeros que participa en el ensamblaje, en el mantenimiento de la integridad estructural y en la regulación de la actividad contráctil de los sarcómeros51. Tiene un peso molecular de aproximadamente 140 kDa, y para cumplir con sus funciones, la cMyBP-C se une a la miosina y a la titina. La unión a la miosina la realiza por medio de dos dominios diferentes, uno en la terminal carboxilo que le sirve para atarse a las colas de las cadenas pesadas y otro en la terminal amino que le sirve para unirse al subsegmento 2 (S2). S2 es la porción de la miosina que conecta la cabeza con la cola.
Normalmente, cuando la cMyBP-C está unida a S2 se reduce la actividad ATPasa de la actinomiosina, pero si ha sido previamente fosforilada por quinasas como la dependiente del complejo Ca2+ /Calmodulina, o la PKA, ese freno se ve disminuido y aumenta la contractilidad miocárdica51,52. Sin embargo, se ha demostrado que las mutaciones en este gen pueden producir tanto cardiomiopatía hipertrófica como dilatada: parece ser que el resultado final dependerá de la sensibilidad al Ca2+ que presente la proteina mutada pues ello determina la fuerza de la contracción y el tiempo medio de relajación50.
En su reciente investigación, Mirza y cols50 estudiaron 8 diferentes mutaciones en genes que codifican para los filamentos delgados TnT, TnC y  -Tm, todas asociadas a cardiomiopatía dilatada y transmitidas de manera autosómica dominante. Los resultados demostraron en todos los casos, una reducción en la sensibilidad al Ca2+ de los filamentos mutados, así como una disminución en la activación de los filamentos delgados. Concluyeron además que, ciertos defectos específicos en la regulación de los miofilamentos representan un estímulo primario para desencadenar la vía de la apoptosis de los miocitos, la dilatación ventricular y su alteración funcional.
-Tm, todas asociadas a cardiomiopatía dilatada y transmitidas de manera autosómica dominante. Los resultados demostraron en todos los casos, una reducción en la sensibilidad al Ca2+ de los filamentos mutados, así como una disminución en la activación de los filamentos delgados. Concluyeron además que, ciertos defectos específicos en la regulación de los miofilamentos representan un estímulo primario para desencadenar la vía de la apoptosis de los miocitos, la dilatación ventricular y su alteración funcional.
Nuevos agentes inotrópicos positivos con efecto sensibilizador al Ca2+ en los miofilamentos
Los agentes inotrópico positivos se caracterizan por mejorar la propiedad contráctil del miocardio entre los que se incluyen los digitálicos, los agonistas adrenérgicos 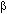 , (dopamia, dobutamina) y los inhibidores de la fosfodiesterasa III (aamrinona, milrinona). El problema con algunos de estos fármacos es que aumentan la incidencia de taquiarritmias ventriculares y de muerte súbita, lo que se debe al aumento intracelular de AMPc, que a su vez incrementa la liberación de Ca2+ del RS. El aumento de Ca2+ intracelular, si bien estimula la contracción, también tiene un efecto cardiotóxico que favorece mecanismos electrofisiológicos que conducen a alteraciones del ritmo así como a la misma apoptosis de los miocitos.
, (dopamia, dobutamina) y los inhibidores de la fosfodiesterasa III (aamrinona, milrinona). El problema con algunos de estos fármacos es que aumentan la incidencia de taquiarritmias ventriculares y de muerte súbita, lo que se debe al aumento intracelular de AMPc, que a su vez incrementa la liberación de Ca2+ del RS. El aumento de Ca2+ intracelular, si bien estimula la contracción, también tiene un efecto cardiotóxico que favorece mecanismos electrofisiológicos que conducen a alteraciones del ritmo así como a la misma apoptosis de los miocitos.
Debido a estos efectos deletéreos,en los últimos años se ha realizado gran cantidad de investigaciones dirigidas al descubrimiento y uso de nuevos fármacos con efecto inotrópico positivo pero con menos efectos adversos. Es así como apareció la familia de los medicamentos conocidos como "sensibilizadores de los miofilamentos al Ca2+", que mejoran la contractilidad pero no directamente a través del aumento del AMPc ni del Ca2+ citosólicos, sino más bien por interacción con algunos de los miofilamentos de los sarcómeros, especialmente con la TnC. De esta manera se evita algunos de los efectos no deseados propios de los fármacos inotrópicos positivos más antiguos53.
Dentro de este nuevo grupo de fármacos sensibilizadores o potenciadores están el levosimendán, el pimobendan, el EMD 57033, el ORG 30029 y el MCI-154. De ellos, el levosimendán se presenta como un fármaco prometedor para el manejo de la insuficiencia del ventrículo izquierdo tanto aguda como crónica54. Propiamente, para producir el efecto sensibilizante, el levosimendán se une a la terminal amino de la TnC cardíaca con gran afinidad y estabiliza la unión del Ca2+ con esta subunidad reguladora (TnC-Ca2+)55,56. La estabilización es dependiente de la concentración citosólica del mismo Ca2+ lo que hace que, a diferencia de otros sensibilizadores, el levosimendán se una y estabilice el complejo TnC-Ca2+ solo durante la sístole y que la relajación del miocardio no se vea afectada57. Además del efecto descrito, el levosimendán favorece la apertura de canales de potasio dependientes de ATP (canales KATP) en los miocitos cardíacos y lisos, lo cual produce vasodilatación y efectos antiarrítmicos. La vasodilatación ha sido reportada en varios lechos vasculares tales como el coronario, pulmonar, renal, esplácnico, cerebral y en las venas sistémicas54.
Conclusiones
En el miocardio,las modificaciones en la concentración citosólica del Ca2+ ionizado determina, de manera directa o indirecta, diversas funciones esenciales como son la génesis y el desarrollo de los potenciales de acción, la regulación de la conducción de los impulsos así como la misma contracción, la integridad celular y la expresión genética, el crecimiento y desarrollo de los miocitos. Por esta razón, las alteraciones en los procesos involucrados en el manejo intracelular del calcio suelen acompañarse de diversas manifestaciones patológicas. Conforme se han ido dilucidando los mecanismos celulares y moleculares en el miocito en los que el calcio intracelular juega un papel importante, también se han entendido mejor los mecanismos fisiopatológicos que explican una serie de cardiopatías. La comprensión de estos mecanismos es esencial para el personal médico que debe llevar a cabo el seguimiento y el manejo de las personas con insuficiencia cardíaca, ciertas arritmias ventriculares, la miocardiopatía arritmogénica del ventrículo derecho, entre otras.
Referencias
1.Roman LM.Signal transduction.En:Boron WF,Boulpaep EL,ed.Medical Physiology. Philadelphia: W.B. Saunders Co.,2002:87-114. [ Links ]
2.Gomperts BD,Kramer IM,Tatham PE.Signal transduction.San Diego:Elsevier Academic Press,2004:145-69. [ Links ]
3.Alberts B,Johnson A,Lewis J,Raff M,Roberts K,Walter P.Molecular Biology of the Cell.New York:Garland Publishing,2002:831-906. [ Links ]
4.Bodi I,Mikala G,Koch SE,Akhter SA,Schwartz A.The L-type calcium channel in the heart:the beat goes on.J Clin Invest 2005;115:3306-17. [ Links ]
5.Molkentin JD.Dichotomy of Ca2+in the heart:contraction versus intracellular signaling.J Clin Invest 2006;116:623-6. [ Links ]
6.Woodcock EA,Matkovich SJ.Cardiomyocytes structure,function and associated pathologies.Int J Biochem Cell Biol 2005;37:1746-51. [ Links ]
7.Gartner LP,Hiatt JL.Texto Atlas de Histología.México D.F.:McGraw-Hill Interamericana,2002:170-173. [ Links ]
8.Hernández A.Mecanismos de transporte 2.Canales proteicos.En:Drucker R,ed. Fisiología médica.México D.F.:Manual Moderno,2005:63-83. [ Links ]
9.Opie LH.The Heart.Physiology,from Cell to Circulation.Philadelphia:Lippincott-Raven Publishers, 1998:43-68. [ Links ]
10.Murphy RA.Contractile mechanism of muscle cells.En:Berne RM,Levy MN.eds. St. Louis: Mosby,1998:269-81. [ Links ]
11.Birkeland JA,Sejersted OM,Toraldsen T,Sjaastad I.EC-coupling in normal and failing hearts.Scand Cardiovasc J 2005;39:13-23. [ Links ]
12.Tayr Y,Frishman WH.The cardiac ryanodyne receptor (RyR2)and its role in heart disease.Cardiol Rev 2005;13:142-6. [ Links ]
13.Kobayashi T,Solaro J.Calcium,thin filaments,and the integrative biology of cardiac contractility.Annu Rev Physiol 2005;67:39-67. [ Links ]
14.Wehrens XH,Lehnart SE,Marks AR.Intracelular calcium release and cardiac disease.Annu Rev 2005;67:69-98. [ Links ]
15.Wu X,Zhang T,Bossuyt J,et al.Local InsP3-dependent perinuclear Ca2+signaling in cardiac myocyte excitation-transcription coupling.J Clin Invest 2006;116:675-82. [ Links ]
16.Nelson DL,Cox MM.Lehninger Principles of Biochemistry.New York:Worth Publishers,2000:1072-1117. [ Links ]
17.Marcus FI,Fontaine GH,Guiraudon G,et al.Right ventricular dysplasia:a report of 24 adult cases.Circulation 1982;65:384-98. [ Links ]
18.Nava A,Martini B,Thiene G,et al.Arrhythmogenic right ventricular dysplasia. Study of selected population.G Ital Cardiol 1988:18:2-9. [ Links ]
19.Norman M,Simpson M,Mogensen J,et al.Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy.Circulation 2005;112: 636-42. [ Links ]
20.Tomé MT,García-Pinilla JM,McKenna W.Update in arrhythmogenic right ventricular cardiomyopathy:genetic,clinical presentation and risk stratificaction. Rev Esp Cardio 2004;57:757-67. [ Links ]
21.Rampazzo A,Nava A,Danieli GA,et al.The gene for arrhythmogenic reight ventricular cardiomyopathy maps to chromosome 14q23-q24.Hum Mol Genet 1994;3:959-62. [ Links ]
22.Alcalai R,Metzger S,Rosenheck S,Meiner V,Chajek-Shaul T.A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia,skin disorder, and wolly hair.J Am Coll Cardiol 2003;42:319-27. [ Links ]
23.Corrado D,Basso C,Thiene G,et al.Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia:a multicenter study.J Am Coll Cardiol 1997;15:1512-20. [ Links ]
24.Dalal D,Nasir K,Bomma C,et al.Arrhythmogenic right ventricular dysplasia.A United States Experience.Circulation 2005;112:3823-32. [ Links ]
25.Yin T,Green KJ.Regulation of desmosome assembly and adhesion.Sem Cell Dev Biol 2004;15:665-77. [ Links ]
26.Mallat Z,Tedgui A,Fontaliran F,Frank R,Durigon M,Fontaine G.Evidence of apoptosis in arrhythmogenic right ventricular dysplasia.N Engl J Med 1996;335: 1190-6. [ Links ]
27.Fontaine G,Mallat Z,Fornes P,Fontaliran F,Frank R.Etiopathogenesis of arrhythmogenic right ventricular dysplasia.Ann Cardiol Angeiol 2000;49:37-47. [ Links ]
28.Yamaji K,Fujimoto S,Ikeda Y,et al.Apoptotic myocardial cell death in the setting of arrhythmogenic right ventricular cardiomyopathy.Acta Cardiol 2005;60:465-70. [ Links ]
29.Matolweni LO,Bardien S,Rebello G,et al.Arrhythmogenic right ventricular cardiomyopathy type 6 (ARVC6):support for the locus assignment,narrowing of the critical region and mutation screening of three candidate genes.BMC Med Genet 2006;7:29-44. [ Links ]
30.Pilichou K,Nava A,Basso C,et al.Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy.Circulation 2006;113: 1171-9. [ Links ]
31.Danieli GA,Rampazzo A.Genetics of arrhythmogenic right ventricular cardiomyopathy.Curr Opin Cardiol 2002;17:218-21. [ Links ]
32.Priori S,Napolitano C.Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+release channels.J Clin Invest 2005 [ Links ]
33.Smith J,Rousseau E,Meissner G.Calmodulin modulation of single sarcoplasmic reticulum Ca2+channels from cardiac and skeletal muscle.Cir Res 1989;64: 352-9. [ Links ]
34.Fruen BR,Bardy JM,Byrem TM,Straburg GM,Louis CF.Differential Ca 2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am J Physiol,Cell Physiol 2000;279:C724-33. [ Links ]
35.Timerman AP,Ogunbumni E,Freund E,Wiederrecht G,Marks AR,Fleischer S. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506- binding protein.Dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplásmico reticulum.J Biol Chem 1993; 268:22992-99. [ Links ]
36.Tinerman AP,Onoue H,Xin HB,et al.Selective binding of FKBP12.6 by the cardiac ryanodine receptor.J Biol Chem 1996;271:20385-91. [ Links ]
37.Weherens XHT,Lehnart SE,Reiken SR,et al.Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2.Science 2004;304: 292-6. [ Links ]
38.Marx SO,Reiken S,Hisamatsu Y,et al.PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor):defective regulation in failing hearts.Cell 2000;101:365-76. [ Links ]
39.Coumel P,Fidelle J,Lucet V,Attuel P,Bouvtain Y.Catecholaminergic-induced severe ventricular arrhythmias with Adams Stokes syndrome in children:report of four cases.Br Heart J 1978;40:28-37. [ Links ]
40.Kontula K,Laitinen PJ,Lehtonen A,Toivonen L,Viitasalo M,Swan H. Catecholaminergic polymorphic ventricular tachycardia:recent mechanistic insights.Cardiovasc Res 2005;67:379-87. [ Links ]
41.Farr MA,Basson C.Sparking the failing heart.N Engl J Med 2004;351:185-7. [ Links ]
42.Baig MK,Goldman JH,Caforio AL,Coonar AS,Keeling PJ,McKenna WJ.Familial dilated cardiomyopathy:cardiac abnormalities are cimmin in asymptomatic relatives and may represent early disease.J Am Coll Cardiol 1998;31:195-201. [ Links ]
43.Grunig E,Tasman JA,Kucherer H,Franz W,Kubler W,Katus HA.Frequency and phenotypes of familial dilated cardiomyopathy.J Am Coll Cardiol 1998;31:186-194. [ Links ]
44.Okazaki T,Honjo T.Payhogenic roles of cardiac autoantibodies in dilated cardiomyopathy.Trends Mol Med 2005;11:322-6. [ Links ]
45.Li D,Tapscoft T,González O,et al.Desmin mutation responsible for idiopathic dilated cardiomyopathy.Circulation 1999;3:461-4. [ Links ]
46.Kamisago M,Sharma SD,DePalma SR,et al.Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy.N Engl J Med 2000;343:1688-96. [ Links ]
47.Schmitt JP,Kamisago M,Asahi M,et al.Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban.Science 2003;299:1410-3. [ Links ]
48.Haghighi K,Gregory KN,Kranias EG.Sarcoplasmic reticulum Ca-ATPase-phospholamban interactions and dilated cardiomyopathy.Biochem Biophys Res Commun 2004;322:1214-22. [ Links ]
49.Chang AN,Harada K,Ackerman MJ,Potter JD.Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in á -tropomyosin.J Biol Chem 2005;280:34343-9. [ Links ]
50.Mirza M,Marston S,Willott R,et al.Dilated cardiomyopathy mutatios in three thin filament regulatory proteins result in a common functional phenotype.J Biol Chem 2005;280:28498-506. [ Links ]
51.Flashman E,Redwood Ch,Moodman-Smook J,Watkins H.Cardiac myosin binding protein C.Its role in physiology and disease.Circ Res 2004;94:1279-89. [ Links ]
52.Ababou A,Gautel M,Pfuhl M.Disecting the N-terminal myosin binding site of human cardiac myosin binding protein C:structure and myosin binding of domain C2.J Biol Chem 2006.In press. [ Links ]
53.Endoch M.Mechanisms of action of novel cardiotonic agents.J Cardiovasc Pharmacol 2002;40:323-38. [ Links ]
54.Toller WG,Stranz C.Lesosimendan,a new inotropic and vasodilator agent. Anesthesiology 2006;104:556-69. [ Links ]
55.Haikala H,Kaivola J,Nissenen E,Wall P,Levyoki J,Lenden IB.Cardiac troponin C as a target protein for a novel calcium sensitizing drug,levosimendan.J Mol Cell Cardiol 1995;27:1859-66. [ Links ]
56.Sorsa T,Pollesello V,Rosevcar PR,Drakenberg T,Kilpelainen I.Stereoselective binding of levosimendan to cardiac troponin C causes Ca2+-sensitizing.Eur J Pharmacol 2004;486:1-8. [ Links ]
57.Gheorghiade M,Teerlink JR,Mebazaa A.Pharmacology of new agents for acute heart failure syndromes.Am J Cardiol 2005;96:68G-73G. [ Links ]
Correspondencia: a Guido Ulate Montero, Apdo 1300-2050, San Pedro, Costa Rica, E-mail:gulate@cariari.ucr.ac.cr, Profesor Catedrático del Departamento de Fisiología de la Escuela de Medicina, Universidad de Costa Rica.
b Estudiante de la Carrera de Medicina, Escuela de Medicina, Universidad de Costa Rica.

