










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Cardiología
Print version ISSN 1409-4142
Rev. costarric. cardiol vol.6 n.3 San José Sep. 2004
Síndrome de Marfán.
Dr. Carlos Mas Romero *, Dr. Roberto Iturrino Monge **
Definición
El Síndrome de Marfán (SM) es una enfermedad del tejido conectivo caracterizado por una herencia autosómica dominante, con una amplia variabilidad clínica. Es el resultado de una mutación en el gen que codifica la fibrilina en el cromosoma 15.
La frecuencia en EEUU es de aproximadamente 1/3000-10000 personas.
Este síndrome se caracteriza por producir manifestaciones oculares, esqueléticas, dérmicas y cardiovasculares, estas últimas con la mayor mortalidad, pueden estar presentes al nacimiento, manifestarse en la infancia en un 25% , y progresar en 1/3 de estos. 1-3
Fisiopatología
Las características clínicas se presentan como resultado del debilitamiento de los tejidos de soporte, por defectos en el gen FBN1, que se ubica en el cromosoma 15q21. 1, y codifica para la proteína defectuosa fibrilina 1, la cual es el principal componente de la matriz de la microfibrilla extracelular. Se produce entonces una degeneración de la fibra elástica, lo cual genera una disminución en la distensibilidad aórtica en respuesta a la onda de presión de pulso o aumento en la rigidez, lo cual se evidencia por ecocardiograma o RMN.
La aorta normal se dilata gradualmente con la edad, sin embargo en el caso del SM, son más notables y más rápidos los cambios.
Desde 1970 se sugirió que la disminución en el impulso de eyección aórtico con beta-bloqueadores podría disminuir el riesgo de disección aórtica en pacientes con SM. Estudios recientes muestran que el propanolol, atenolol o metoprolol tienen un efecto heterogéneo, aumentan la distensibilidad aórtica y disminuyen su rigidez y la velocidad de la onda de pulso en un subgrupo de pacientes, mientras que en los que no responden, ocurre un deterioro en la distensibilidad aórtica 2 , de significado clínico aún no claro. Su beneficio está en sus propiedades inotrópicas y cronotrópicas negativas, más que en su efecto sobre la presión arterial. La respuesta a los beta-bloqueadores es mejor en pacientes más jóvenes con aortas más pequeñas (menores de 40mm). 4, 5, 6 Recientemente se ha empezado a utilizar calcio antagonistas, mostrando buenos resultados en niños, aunque su uso aún no es recomendado por la Asociación Americana de Pediatría. 1, 2
Diagnóstico
La enfermedad se presenta típicamente con dilatación aórtica progresiva asociada con incompetencia de la válvula aórtica, prolapso e incompetencia de la válvula mitral, dislocación lenticular y miopía, y un cuerpo alto y delgado.
El diagnóstico debe hacerse con base en un abordaje multidisciplinario (cardiología, genética, cirugía cardiovascular, oftalmología, enfermería y ortopedia). Esta basada en la llamada Nosología Diagnóstica de Ghent (1996), como se muestra en la Tabla 1. 2
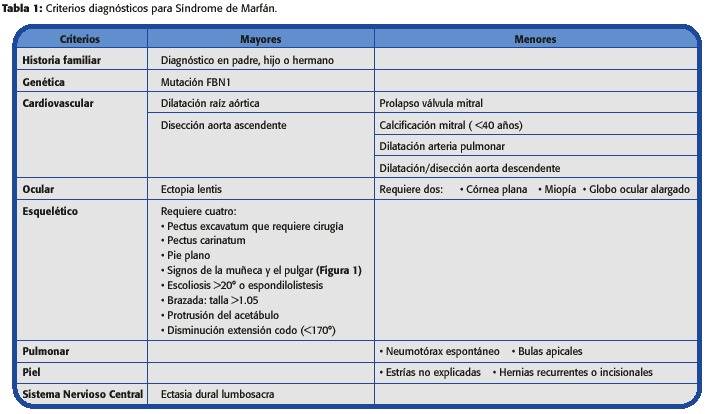
Requiere:
Un criterio mayor en dos sistemas
Un tercer sistema involucrado
En vista de que existen características que aparecen con la edad, pacientes jóvenes con historia familiar de SM que no cumplen con los criterios, o aquellos con características del SM sin historia familiar, que les falta el compromiso de un sistema para el diagnóstico, se les debe someter a evaluaciones seriadas hasta los 18 años.
Evolución clínica
Durante la niñez y adolescencia de los portadores del SM, la disfunción valvular mitral, así como las anormalidades aórticas se desarrollan y progresan gradualmente, a menudo sin síntomas, pero pueden causar morbi-mortalidad importante a finales de la segunda década de la vida, especialmente en algunas pacientes femeninas.
Por ecocardiograma, el prolapso mitral es evidente en 75% de los pacientes portadores de SM, sin embargo sólo 25% de ellos desarrollan regurgitación mitral clínicamente significativa 3.

El pronóstico de los pacientes está determinado por las anormalidades de la raíz aórtica, las cuales predisponen a dilatación y disección progresiva, y pueden llevar a regurgitación aórtica. La disección aórtica se relaciona con el diámetro de la raíz de la aorta. Aquellos pacientes sin dilatación, o cuya raíz mide menos de 50mm rara vez presentan disección aórtica 7.
El 90% de las muertes se deben a patología de la aorta ascendente. La sobrevida de los pacientes no tratados es de 40 años aproximadamente aunque el rango es grande, sin embargo, en los últimos 30 años ha mejorado sustancialmente debido a mejoras en el manejo tanto médico como quirúrgico, y actualmente la expectativa de vida es de 70 años 3. Esto debido a que la utilización de los beta-bloqueadores disminuye la progresión de la dilatación aórtica y el riesgo de disección en algunos pacientes 4. Además la cirugía profiláctica de la raíz aórtica tiene mejores resultados que la de emergencia. Sin embargo, estudios recientes señalan otra causa de muerte en estos pacientes; la dilatación ventricular izquierda también puede causar muerte súbita, secundaria a arritmias ventriculares. 2, 3
Pruebas de Gabinete
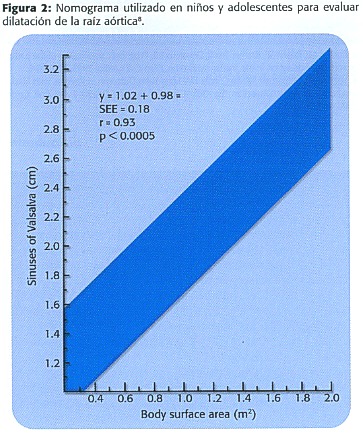
Se requiere para el abordaje correcto, las pruebas que se detallan en la Tabla 2 3.

Complicaciones cardiovasculares
Incluyen: prolapso y regurgitación de la válvula mitral, dilatación ventricular izquierda y falla cardiaca y dilatación de la arteria pulmonar. Sin embargo, la dilatación de la raíz de la aorta es la causa más común de morbi-mortalidad.
Los factores de riesgo para disección aórtica incluyen 2:
Diámetro aórtico mayor a 50mm.
Extensión de la dilatación aórtica mas allá del seno de Valsalva.
Tasa de dilatación aórtica rápida (>5% anual, o >2mm/año).
Historia familiar de disección aórtica.
Manejo médico
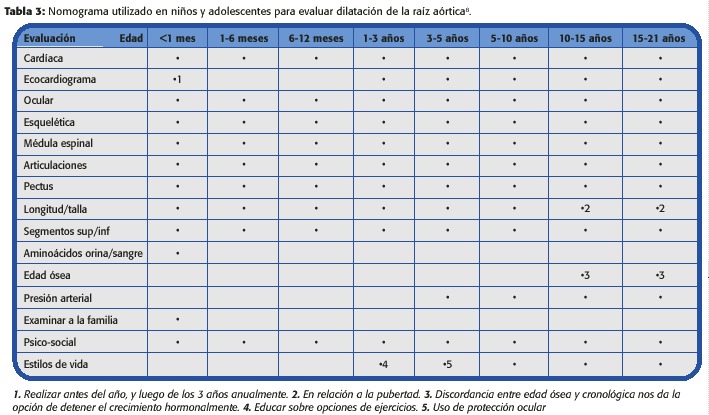
En los pacientes con SM se recomienda un abordaje multidisciplinario durante toda la edad pediátrica. Las guías se presentan en la Tabla 3. 1
Además, las siguientes recomendaciones específicas de manejo cardiovascular son válidas en la infancia y la edad adulta 2, 3:
Al menos una evaluación anual. Debe darse si es posible a nivel multidisciplinario, a cargo de profesionales con experiencia en el campo, y debe incluir
Historia clínica
Examen físico
Ecocardiograma transtorácico anual y dependiendo del diámetro aórtico (mayor de 40mm)y de la tasa de dilatación (más de 2mm por año)se puede hacer cada 6 meses). Evaluar necesidad de RMN, TAC y/o US abdominal en caso necesario 7.
Referencia a cirugía profiláctica de la raíz aórtica cuando el diámetro a nivel del seno de Valsalva exceda 50mm o Holter de 24 horas en pacientes con dilatación del ventrículo izquierdo 9.
En caso de embarazo el riesgo de disección aumenta si el diámetro es >40mm, lo que obliga a un monitoreo frecuente en el embarazo y el puerperio.
Evitar el ejercicio isométrico y máximo, y evitar deportes de contacto (fútbol, baloncesto, boxeo, artes marciales, rugby, clavados, buceo)para evitar complicaciones aórticas o oculares.
Uso de beta-bloqueadores, en todos los pacientes con dilatación aórtica a cualquier edad, principalmente en los más jóvenes. El tratamiento profiláctico es más efectivo con un diámetro aórtico menor a 40mm 4.
Profilaxis de endocarditis en todos los pacientes con SM excepto los que no tienen regurgitación valvular o válvula protésica.
El uso de beta-bloqueadores se ve limitado por su tolerancia y contraindicaciones (asma, falla cardíaca, bradiarritmias), y en cuanto al ejercicio la morbi-mortalidad precipitada se presenta sobretodo en casos de dilatación aórtica importante 2, 3.
Se ha sugerido el uso de inhibidores de la ECA, pero su utilidad no se ha demostrado 2.
Manejo quirúrgico
Para el reemplazo de la raíz aórtica se sugiere el procedimiento de Bentall modificado (reemplazo de válvula y aorta ascendente). Si las cúspides son normales se recomienda la reimplantación de la válvula o la remodelación de la raíz aórtica. Las indicaciones de cirugía se muestran en la Tabla 4 3.

Bibliografía:
1. AAP. Health Supervision for Children with Marfan Syndrome. Pediatrics 1996; 5: 978-982. [ Links ]
2. Dean JC. Management of Marfan Syndrome. Heart 2002; 88: 97-103. [ Links ]
3. Webb GD, David TE. Marfan Syndrome: A cardiovascular perspective. En: Diagnosis and Management of Adult Congenital Heart Disease. 2003: 481-486. [ Links ]
4. Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long term beta adrenergic blockade in Marfan s Syndrome. N Engl J Med 1994; 330: 1335-41. [ Links ]
5. Rossi-Foulkes R, Roman MJ, Rosen SE, et al. Phenotypic Features and Impact of Beta Blocker or Calcium Antagonist Therapy on Aortic Lumen Size in the Marfan Syndrome. Am J Cardiol 1999; 83: 1364-8. [ Links ]
6. Ríos AS, Silber EN, Bavishi N, et al. Effect of Long Term Beta Blockade on Aortic Root Compliance in Patients With Marfan Syndrome. Am Heart J 1999; 137: 1057-6. [ Links ]
7. Kornbluth M, Schnittger I, Eyngorina I, et al. Clinical Outcome in the Marfan Syndrome With Ascending Aortic Dilatation Followed Anually by Echocardiography. Am J Cardiol 1999; 84: 753-5. [ Links ]
8. Roman MJ, Rosen SE, Kramer-Fox R, Devereux RB. Prognostic significance of the pattern of aortic root dilatation in the Marfan syndrome. J Am Coll Cardiol 1993; 22: 1470-6. [ Links ]
9. Yetman AT, Bornemeir RA, McCrindle BW. Long Term Outcome in Patients with Marfan Syndrome: Is Aortic Dissection the Only Cause of Sudden Death?J Am Coll Cardiol 2003; 41: 329-332. [ Links ]
* Residente de Pediatría, Hospital Nacional de Niños Dr. Carlos Sáenz Herrera, C. C. S. S.
** Asistente Especialista Servicio de Cardiología, Hospital Nacional de Niños Dr. Carlos Sáenz Herrera, C. C. S. S. Apartado 12795-1000 San José, e-mail: drmas@doctor.com

