










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Cardiología
Print version ISSN 1409-4142
Rev. costarric. cardiol vol.5 n.3 San José Dec. 2003
Introducción
Existen numerosas publicaciones en las revistas médicas de las diversas áreas de la medicina, tanto de índole clínico como experimental, que sugieren que las llamadas citocinas pro-inflamatorias, en especial el Factor de Necrosis Tumoral alfa (FNT-alfa), juegan un papel muy importante en la patogenia de la insuficiencia cardíaca crónica. El FNT-alfa, que se creía era producido únicamente por células del sistema inmunológico (particularmente monocitos y macrófagos activados), se ha encontrado también que puede sintetizarse en las células miocárdicas (miocitos) del corazón insuficiente, pero no en el normal. En el laboratorio se manejan modelos de corazón con sobrecarga de presión o de volumen, que hacen que el miocardio insuficiente sea capaz de sintetizar estas moléculas, en particular el FNT-alfa. Por esta razón se les llama también citocinas estrés-inducidas. Estas citocinas producen una franca disminución del inotropismo miocárdico, estimulan la síntesis del tejido conectivo intersticial, causan una miocardiopatía e inducen apoptosis, tanto in vivo como in vitro. La expresión de receptores específicos para estas citocinas se encuentran elevadas en la insuficiencia cardíaca crónica.
Estas biomoléculas son capaces por sí mismas de producir un síndrome de insuficiencia contráctil del miocardio y el FNT-alfa (también conocido como caquectina) se cree que es causante en gran parte de la caquexia observada en los cardiópatas terminales. Dado que los niveles del FNT-alfa están en relación con la severidad de la insuficiencia cardíaca, se considera que estas moléculas son mediadores importantes en la progresión de la enfermedad. La obtención de compuestos químicos que contrarresten estas citocinas, podrían ser de gran utilidad en el manejo de este síndrome congestivo.
Corazón e Inflamación
Desde hace más de 200 años se sabe que el corazón puede ser asiento de procesos infecciosos e inflamatorios en sus diversos componentes anatómicos: pericardio, miocardio, endocardio y válvulas. Pero no fue si no a comienzos de los años 90s del recién pasado siglo, en que por primera vez se demuestra que la placa ateromatosa, lesión primordial de la aterosclerosis, puede presentar un componente inflamatorio desde el punto de vista histopatológico (37, 47, 48). Estos primeros estudios se hicieron en arterias coronarias. También se descubrió que durante el proceso evolutivo de la aterosclerosis hay un componente inflamatorio crónico sistémico de baja intensidad (64, 68, 79, 90, 123, 124, 126). Cuando dicho proceso inflamatorio se incrementa, es capaz de provocar cambios en la placa ateromatosa que aumenta su vulnerabilidad, causando la ruptura, que es la culpable en gran parte, de los síndromes coronarios agudos (79, 90, 123, 124, 125). Este proceso inflamatorio de baja intensidad se hace evidente el encontrarse que la Proteína C Reactiva (PCR), que es un marcador sensible de inflamación, se encuentra elevada moderadamente, siendo un percusor importante de un evento coronario agudo, cuando sus niveles séricos se incrementan (95, 105, 121, 122). En dichas investigaciones se usó la PCR sensibilizada.
Por muchos años se creyó que el desarrollo de la placa ateromatosa era estrictamente un proceso degenerativo cuyos agentes causales eran los ya conocidos factores de riesgo coronario. Nunca antes se pensó que hubiese en su génesis un componente inflamatorio.
Para sorpresa aún mayor de la comunidad médica, se descubrió que varios microorganismos como el Helicobacter pylori, la Chlamidia pneumoniae, los citomegalovirus y los miembros del grupo de virus del herpes, tenían alguna participación en el desarrollo de la placa ateromatosa. Este hecho plantea la interrogante de que cierto grupo de antibióticos, especialmente macrólidos de la última generación (azitromicina) pudieran ser de alguna utilidad en el tratamiento de los síndromes coronarios agudos. No es objetivo de esta revisión hacer un escrutinio detallado de la evidencia al respecto.
También a comienzos de la década de los 90s del siglo pasado se empezó a sospechar que en la insuficiencia cardíaca crónica podrían existir los componentes inflamatorios. Con este pensamiento Pye y cols. (21) en un estudio de investigación clínica en pacientes con insuficiencia cardíaca crónica, encontraron niveles séricos altos de PCR y que había una relación entre la gravedad del padecimiento y las cifras de este marcador de inflamación.
Con el mismo propósito se publicó un trabajo de investigación clínica orientado a medir la eritrosedimentación (ESM) en individuos con insuficiencia cardíaca crónica. En dicho estudio se encontró que en los casos leves a moderados la ESM se encontraba muy aumentada, pero en las formas moderadas-severas, era normal o aún baja (25). Este es un hallazgo aparentemente paradójico.
Insuficiencia Cardíaca y Citocinas
En 1975, Carswell y cols. (1) descubrieron el factor de necrosis tumoral-alfa (FNT-alfa), al encontrar en ratones a los que se les había inyectado endotoxina, un compuesto que, al inyectarlo a otros roedores afectos por un estirpe tumoral determinada provocaba la necrosis del tumor. De allí el nombre de FNT con que se le llamó. Este compuesto resultó ser una citocina con múltiples actividades biológicas.
Las citocinas, que juegan un papel muy importante en la respuesta inflamatoria inmunológicamente inducida, son biomoléculas muy pequeñas (15 a 30 kDa), de naturaleza proteica. Son secretadas por células que pertenecen al sistema inmunológico, principalmente monocitos y macrófagos activos, en respuesta a varios estímulos inductores, entre ellos los causados por microorganismos (9, 10, 15, 20).
Varios estudios de investigación experimental habían puesto de manifiesto que el FNT-alfa tenía, entre algunas de sus características biológicas, marcados efectos inotrópicos negativos en el corazón sano, y que podría ser el factor que causaba la insuficiencia cardíaca severa observada en el choque endotóxico causado por bacterias Gram negativas (4, 5, 6, 8, 10, 12, 15, 26, 36, 42, 49, 52), pero fueron Levine y cols. del grupo de investigación de Milton Packer, del Centro Médico Monte Sinai, de Nueva York, quienes en un trabajo de investigación clínica monumental demuestran, por vez primera, que en los pacientes con insuficiencia cardíaca crónica se encuentran niveles elevados del FNT-alfa, siendo los títulos tanto más altos cuando más severa es la insuficiencia (19). Este estudio, ya un clásico en el área de investigación de las citocinas, provocó un enorme interés en esa área, lo que hizo que se iniciaran muchos estudios experimentales como clínicos, sobre esta interesante biomolécula que pertenece al grupo de las citocinas pro-inflamatorias (3, 4, 5, 6, 8, 10, 12, 15, 54, 56). Entre el grupo de citocinas pro-inflamatorias estudiadas, se encontró que la Interleucina-1 (IL-1) y la Interleucina-6 (IL-6) tenían participación en el síndrome de insuficiencia cardíaca crónica (9, 13, 16, 29, 42, 43, 50, 61, 72, 76). Últimamente se ha reportado que la Interleucina-18 (IL-18), biológicamente relacionada a la IL-1, también participa en dicho síndrome (92, 101, 108). El FNT-alfa y la IL-6 parecen ser las más activas, o por lo menos han sido las más estudiadas. (9, 11, 19, 61, 63).
Hemos mencionado que estas citocinas pro-inflamatorias eran sintetizadas y secretadas por moléculas del sistema inmunológico, particularmente monocitos y macrófagos activados, en respuesta a varios tipos de estímulos inductores, frecuentemente de tipo infeccioso. Sin embargo, en 1995 se publicaron varios trabajos de investigación muy bien diseñados, en los que se descubrió que algunas células nucleares del corazón, particularmente los miocitos, eran capaces de sintetizar y secretar estas citocinas, principalmente el FNT-alfa, en respuesta a varios inductores de tipo mecánico, como sobrecargas volumétricas o de presión (42, 54, 72). Por esa razón, algunos investigadores las llaman también citocinas estrés-inducidas (41, 49, 54).
Este descubrimiento estremeció el edificio de la Inmunología, que establecía, casi como un dogma de fe, que los componentes de la respuesta inmunológica, como las citocinas, únicamente podrían ser secretadas por dichas células y no por otras, como son los miocitos.
Otro importante hallazgo fue que los miocitos sanos, sometidos a ningún tipo de estrés, tanto mecánico como infeccioso, eran incapaces de sintetizar dichas citocinas. Los numerosos estudios de investigación, experimentales como clínicos, han encontrado que las citocinas inflamatorias tienen muchos efectos sobre el aparato cardiovascular. Entre ellos tenemos: la insuficiencia cardíaca, edema agudo de pulmón, caquexia cardíaca, miocardiopatía, desacoplamiento del receptor beta de la adenilciclasa (el segundo mensajero), activación del sistema genético para promover su síntesis, disfunción endotelial y apoptosis (3, 4, 5, 6, 7, 8, 9, 10, 12, 13, 16, 26, 30, 50, 54, 55, 56, 57, 66, 71, 84, 85). Los primeros estudios que estas citocinas estuvieron orientados a la investigación de la patogenia del choque endotóxico, especialmente del FNT-alfa. La caquectina, relacionada con la caquexia observada en el cáncer terminal, resultó ser el mismo FNT-alfa, que tiene también una participación importante en la caquexia del cardíaco en su fase terminal (4, 8, 22, 67, 92).
Al ser sintetizadas y secretadas por un miocito "enfermo" y no por las células del sistema inmunológico, se plantea la duda de que si a esta respuesta se le puede llamar con toda propiedad una "respuesta inmunológica", a pesar que estas biomoléculas pertenecen al grupo de los "efectores" de dicho sistema. Existe pues una controversia al respecto.
Las citocinas "estrés inducidas" tienen muchos efectos biológicos comunes con el llamado "fenotipo" de la insuficiencia cardíaca. Esto hizo que se presumiera que estas moléculas biológicamente muy activas jugaran un papel decisivo en la insuficiencia cardíaca crónica (41, 56, 60, 65, 93, 101). Nace así la "hipótesis de las citocinas" (54, 119), y se plantea la interrogante de que estas biomoléculas podrían tener alguna participación en la progresión o deterioro de la insuficiencia cardíaca. Esta "hipótesis de las citocinas" no significa que estas moléculas sean la causa de la insuficiencia cardíaca, sino que su papel está en relación con la progresión clínica del síndrome de falla contráctil del corazón. Un hecho muy interesante es que no existe una diferencia en la expresión de las citocinas pro-inflamatorias, tanto en la disfunción causada por una miocardiopatía dilatada primaria como en la fase dilatada de la insuficiencia cardíaca de la cardiopatía isquémica (101). También muy interesante es el hecho de que la expresión del FNT-alfa es independiente de que el tipo de sobrecarga cardíaca sea volumétrica o de presión.
La mayoría de los efectos biológicos de las citocinas son mediados a través del mecanismo "agonista/receptor". Por lo tanto para involucrar a estas bio-moléculas en la patogénesis de la insuficiencia cardíaca crónica, se tenía que demostrar la existencia de receptores miocárdicos para estas citocinas pro-inflamatorias. Estos receptores fueron detectados en las membranas de los miocitos enfermos (52, 53, 57, 69, 74, 75, 78, 81, 87, 93, 101, 109, 117, 119). Se han descrito dos receptores del FNT-alfa: el receptor 1 del FNT-alfa (E1-FNT-alfa) y el receptor 2 del FNT-alfa (E2-FNT-alfa). Los dos receptores se encuentran en proporciones similares en el miocardio normal (52, 53, 93, 101). Sin embargo, los efectos biológicos de cada receptor son distintos, teniendo ambos igual afinidad de fijación para el FNT-alfa. El efecto inotrópico negativo es mediado por la interacción del FNT-alfa con el R1-FNT-alfa (53, 93, 101).
Los complejos citocina-receptor del miocardio se desprenden, formando los llamados complejos solubles del receptor, que circulan libremente en el compartimiento vascular. Al desprenderse, ligan las citocinas en forma continua, contribuyendo así a mantener un equilibrio o modulación en el sistema (Figura 1).
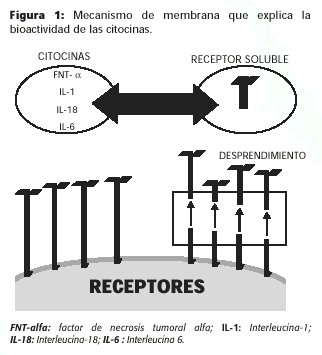
Con toda la información científica, tanto a nivel clínico como experimental, se demuestra que las citocinas pro-inflamatorias juegan un papel importante en la remodelación del ventrículo izquierdo (dilatación), que llevan al paciente a la fase terminal de su padecimiento (Figura 2).
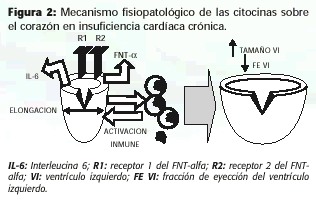
Además de los efectos inotrópicos negativos de estas citocinas, hay también otros efectos nocivos sobre el miocardio, como lo hemos mencionado. Otro de los mecanismos que causan el remodelado (dilatación de las cámaras cardíacas), es el daño sobre la matriz extracelular. Dicho daño consiste en la degradación progresiva de la matriz por la activación de las metaloproteínasas de la matriz (MPMs) inducidas principalmente por el FNT-alfa (110, 119). La "digestión" de la malla de colágeno fibrilar le quita soporte a los miocitos y favorece así la dilatación ventricular.
Ridker (125) ha acuñado el concepto del "suelo común" (common soil, en inglés) para explicar la multiplicidad de factores que intervienen en el proceso inflamatorio del corazón y sus relaciones con la diabetes mellitus tipo II. Al presente ya se visualizan las "puntas de los icebergs" pero aún no conocemos en "profundidad" las conexiones precisas de estos mecanismos a nivel celular y molecular.
Progresión de la Insuficiencia Cardíaca Crónica
Se ha observado que durante varios años la insuficiencia cardíaca se mantenía controlada en forma más o menos estable con el esquema terapéutico clásico de digital y diurético, pero que súbitamente y sin causa aparente, se presentaba un deterioro de la condición hemodinámica que rápidamente llevaba al paciente a la muerte, ya por falla mecánica o por una arritmia maligna (75, 101, 126). Esta progresión de la enfermedad se asocia a un franco incremento en la actividad del eje neuro-endocrino, particularmente un aumento en los niveles séricos de angiotensina II, norepinefrina y Endotelina-1(E-1), así como también una elevación en la actividad de la arginina-vasopresina (hormona antidiurética) (58, 75, 101, 103, 106). La sospecha de que el incremento en los niveles séricos de estas hormonas estuviese estrechamente relacionado al deterioro de la insuficiencia cardíaca crónica fue corroborado cuando al tratar a estos pacientes con inhibidores de la enzima convertidora o convertasa (IECAs) y bloqueadores beta-adrenérgicos se observaba una notable mejoría de los síntomas clínicos, disminuyendo ambién la morbi-mortalidad de la enfermedad (31, 39, 86, 94, 96, 112, 113, 114, 118). Numerosos trabajos en tal sentido, tanto experimentales como clínicos, se llevaron a cabo en varios países. La creciente disponibilidad de fármacos antagonistas neuro-hormonales ha transformado la terapia de la insuficiencia cardíaca de un esquema básicamente paliativo (digital/diuréticos) a uno que modifica la evolución de la enfermedad (75). Pero esa no fue toda la historia: se pensó que había otros factores químicos que podrían explicar ese deterioro de la función miocárdica y se empezó a explorar la posibilidad de que ciertas bio-moléculas cardiodepresoras como el FNT-alfa, podrían también producirse en la insuficiencia cardíaca crónica y acelerar el deterioro clínico. (27, 29, 36, 42, 43, 52, 101, 126).
Revisemos brevemente algunos mecanismos que actúan en la génesis de la insuficiencia cardíaca crónica en su fase terminal. La vasoconstricción sistémica es un importante mecanismo compensatorio en la insuficiencia cardíaca. Esta respuesta vasoconstrictora depende de la integridad del sistema nervioso simpato-adrenal y del eje renina-angiotensina-aldosterona (58). Este mecanismo provoca una importante disfunción endotelial, demostrada en numerosos trabajos clínicos y experimentales, que es en definitiva el causante mayor de la vasoconstricción (24, 27, 30, 40, 43, 46, 58, 73, 76, 83, 100, 102, 106, 107). Además de la alteración de la vasodilatación endotelio-dependiente, el aumento en la circulación de la E-1 que se observa en la insuficiencia cardíaca, puede jugar también un papel importante en dicha vasoconstricción.
Se ha visto que los cambios en la E-1 plasmática están relacionados a incrementos en las otras neurohormonas como la vasopresina y la norepinefrina que estimulan in-vitro la liberación de endotelina (27, 43, 94). Además, el estrés inducido por los cambios hemodinámicos llevan también a un rápido aumento en la circulación plasmática de los niveles de la E-1. La principal fuente de E-1 es el lecho vascular pulmonar, y su secreción aumenta en relación a la severidad de la insuficiencia cardíaca. Esta endotelina es la causante del importante aumento de la resistencia vascular pulmonar en la insuficiencia cardíaca (43). El marcado aumento de los vasoconstrictores derivados del endotelio parece ser una consecuencia, mas que una causa de la insuficiencia cardíaca (58, 101). En realidad la vasoconstricción es uno de los más importantes mecanismos compensatorios que ocurren en la insuficiencia cardíaca, y descansa grandemente en hormonas tales como la vasopresina y la norepinefrina que inducen la producción de endotelina (45). Además la norepinefrina de por sí es un vasoconstrictor a través de su mecanismo alfa-agonista adrenérgico. Este aumento consecuente de la post-carga explica la disminución del débito cardíaco en un corazón ya deprimido.
Las citocinas pro-inflamatorias, en especial el FNT-alfa, tienen aparte de su efecto inotropo-negativo, un efecto inductor de la disfunción endotelial, con el consiguiente incremento en la respuesta vasoconstrictora. Además, el eje neurohormonal incrementa la síntesis y liberación de las citocinas pro-inflamatorias (102, 103). Vemos pues, que el estrés hemodinámico estimula la producción de estas citocinas (de allí el nombre de estrés–inducidas, que también reciben) y también existe una interacción citocinas/eje neurohormonal en la modulación de dicha respuesta (102, 103).
Los síntomas limitantes de la insuficiencia cardíaca incluyen fatiga, que es una expresión de bajo gasto cardíaco, y disnea, provocada por la congestión pulmonar pasiva crónica. La fatiga marcada, que a veces llega a ser invalidante, también se explica por la marcada vasoconstricción que lleva a una pobre perfusión tisular (58, 59). Esta condición de bajo gasto con hipoperfusión tisular, en particular del tejido muscular, que se observa en la insuficiencia cardíaca crónica, ha llevado a Andrew J. S. Coats a formular la "hipótesis muscular" de la insuficiencia cardíaca crónica.
Probablemente los mensajes a nivel molecular que conducen a este deterioro en la insuficiencia cardíaca se originen en el área tisular y promuevan a una respuesta vasoconstrictora aún mayor, transformando la respuesta al inicio compensatoria en una respuesta deletérea al final, ya que compromete aún más la condición metabólica tisular. Este mecanismo también explicaría la progresión clínica de la enfermedad. De hecho, en esta etapa, la fatiga muscular se vuelve mas intensa y el gasto cardíaco disminuye aún más. El correlato farmacológico corrobora esta situación: el paciente mejora con los fármacos inhibidores de la convertasa y los bloqueadores beta-adrenérgicos. Además se demuestra una mejoría de la disfunción endotelial, mejor perfusión tisular y menos disnea/fatiga.
Aunque no hay duda de la interacción eje neuro-hormonal/citocinas pro-inflamatorias, no existe sin embargo una demostración de tal interacción a nivel molecular (101).
Uso de Fármacos Anticitocinas en la insuficiencia Cardíaca Crónica
Ha sido ampliamente documentado que las citocinas pro-inflamatorias deprimen la contractilidad miocárdica y contribuyen a la importante disfunción endotelial observada en la insuficiencia cardíaca crónica, favoreciendo así la progresión (deterioro) de la enfermedad ("hipótesis de las citocinas"). Con esta información, es lógico suponer que si su actividad es antagonizada podemos mejorar la condición hemodinámica del corazón y detener temporalmente su progresión.
Siendo esta respuesta de las citocinas mediada en parte por el sistema inmunológico se propone lo que los entendidos en la materia llaman el "manejo inmunomodulatorio" de la enfermedad (97, 104, 111, 115, 116).
Los trabajos sobre el uso de los inhibidores de la enzima convertidora de angiotensina (IECAs) han demostrado que su uso provoca un descenso de FNT-alfa, probablemente debido a que modifica favorablemente la condición hemodinámica de los pacientes, disminuyendo el factor de estrés mecánico (citocinas estrés–inducidas), aunque no se descarta de que pueda haber una interrelación entre las neurohormonas (norepinefrina, angiotensina) y las citocinas pro-inflamatorias, en particular el FNT-alfa. Por ese o esos mecanismos podríamos decir que los fármacos tipo IECA tienen un efecto anticitocina, directo y/o indirecto.
En 1998, en un estudio publicado en Lancet, Sliwa y colaboradores informan de los efectos benéficos del fármaco pentoxifilina sobre la función ventricular izquierda en pacientes con insuficiencia cardíaca crónica causada por miocardiopatía dilatada idiopática (77). Este fue un estudio pequeño, que incluyó 28 pacientes, en quienes se demostró una mejoría de la fracción de eyección ventricular izquierda medida por ecocardiografía. Auque los resultados son muy buenos se considera que el estudio es muy pequeño y con un diseño experimental un poco laxo como para sacar conclusiones definitivas, sugiriéndose hacer un estudio mayor y con mejores controles. Dos años después, en el 2000, Bozkurt y su grupo, del Centro Médico de la Universidad de Baylor, Texas, presenta los resultados de un estudio abierto en que usaron talidomida en pacientes con insuficiencia cardíaca avanzada y niveles altos del FNT-alfa, con una respuesta clínica aceptable (98). Pero el estudio es también muy pequeño y no permite sacar conclusiones firmes. Estos serían indicios que gozan de cierta solidez, que los fármacos que disminuyen los niveles del FNT-alfa puedan mejorar el pronóstico de este síndrome.
La talidomida y la pentoxifilina se consideran drogas que son inhibidores inespecíficos de las citocinas, al afectar la síntesis de los mediadores inflamatorios bloqueando su activación transcripcional (manipulación genética).
Este mismo grupo, en 1999 (89), habían publicado un pequeño trabajo sobre el uso del etanercept (Enbrel ®, comercialmente) en el que se demostraban un franca mejoría clínica de los pacientes. Ellos investigaron este fármaco después de conocer los resultados que se habían obtenido con este medicamento en individuos que habían recibido endotoxina por vía endovenosa (51). El etanercept se ha usado con éxito en el tratamiento de la artritis reumatoide, llevando a remisiones clínicas. Es un receptor del FNT-alfa, de origen recombinante humano, con una afinidad 100 veces mayor que la de los receptores naturales del FNT-alfa por esta citocina.
Basado en estos estudios preliminares Anker publica un artículo titulado: ¿Ha llegado el momento del uso de la terapia anticitocina en la insuficiencia cardíaca crónica? (104). El autor afirma que hay pruebas suficientes del efecto negativo de estas citocinas sobre la evolución de la enfermedad y que ha llegado la hora en que se diseñen estudios más completas para valorar el uso de fármacos anticitocinas en la insuficiencia cardíaca de larga data.
Se diseñan entonces dos grandes estudios multicéntricos para valorar el uso de etanercept en la insuficiencia cardíaca crónica. Uno de ellos es el RENAISSANCE (acrónimo de "The Randomized Etanercept North AmerIcan Strategy to Study ANtagonism of CytokinEs"), que se desarrolló en Norteamérica. El otro, llamado RECOVER (acrónimo de "Research into Etanercept CytOkine antagonism in VEntRicular Dysfunction"), que se realizó en Europa y Australia. El resumen de estos dos grandes estudios se sintetiza en el RENEWAL (acrónimo de "Randomized EtaNErcept Worldwide EvALuation"). Ambos estudios incluyeron un total de 2.048 pacientes, con un excelente diseño experimental (120). Los puntos finales fueron ingresos hospitalarios y muerte por agravamiento de su insuficiencia cardíaca. Fueron dos estudios muy costosos y al final las conclusiones de los resultados fueron que el etanercept empeoró la condición cardíaca. Al lector que tenga especial interés por los resultados de ambos estudios le sugerimos revisarlos, ellos son: 1) Anker S D, Cotas AJS: How to RECOVER for RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int. J. Cardiol. 2002; 6:123- 32 y 2) Mann DL: Inflammatory Mediators and the Failing Heart. Past, Present, and the Foreseeable Future. Circ. Res. 2002; 91: 988-998 (119, 120). En ambos artículos también se revisan los resultados del estudio ATTACH (acrónimo de "The AnTi-TNF-alfa Therapy Against CHF"). Este fue un estudio pequeño de fase II en insuficiencia cardíaca crónica moderada a avanzada, que estudio a 150 pacientes tratados con infliximab (Remicade®, comercialmente). Este es un fármaco del tipo anticuerpo monoclonal contra el FNT-alfa, ya usado con éxito en la enfermedad de Crohn. En este estudio se demostró la ineficacia de dicho fármaco en la insuficiencia cardíaca, ya que no solo tuvo un efecto beneficioso nulo, sino que empeoró la condición clínica de los pacientes. Ante estos resultados Anker y Coats (120) se preguntan: ¿Ha muerto la hipótesis de las citocinas? Mann (119) se hace igual cuestionamiento.
Sin embargo, ellos mismos afirman que es tan convincente el efecto negativo cardiovascular que tienen las citocinas pro-inflamatorias que es prematuro descartar el uso de fármacos anticitocinas en esta condición clínica. Probablemente habrá que hacer una mejor selección de los pacientes y estudiar nuevas dosis. Podría ser que dichos fármacos puedan tener efectos colaterales tóxicos sobre el corazón, independientemente de su acción anticitocina, sobre todo teniendo en cuenta que actúan a nivel de membrana celular. Estos reconocidos autores piensan que es muy prematuro proclamar: " ha muerto el rey, larga vida al rey".
Conclusión
La "hipótesis de las citocinas" ha abierto una nueva ruta en el conocimiento del papel de las citocinas pro-inflamatorias en la patogénesis de la insuficiencia cardíaca. Hay que continuar con los estudios de los fármacos anticitocinas. Es probable que en el futuro cercano, estas drogas ocupen un lugar de privilegio en el manejo de la insuficiencia cardíaca como el que ahora tienen los IECAs y los bloqueadores beta.
Referencias
1. Carswell EA, Old IJ, Kassel RI, Et al: An endotoxin induced serum that causes necrosis of tumors. Proc Nat Acad Sci USA. 1975: 72: 234-248 [ Links ]
2. Cohn JN, Levine TB, Olivari MT, et al: plasma norepinephrine as a guide to prognosis in patients with chronic congestive hert failure. N Engl J Med. 1984; 311: 819-823. [ Links ]
3. Sugarman BJ, Aggarwal BB, Hass PE, et al: Recombinant hunan necrosis factor-alpha:effects on proliferation of normal and transformed cells in vitro. Science. 1985; 230: 943-945. [ Links ]
4. Beutler B, Milsark IW, Cerami AC: passive inmunization against cachectjn/tumor necrosis factor protecs mice from lethal effect of endotoxina. Science. 1985; 229:869-871. [ Links ]
5. Hariri RJ, Fahey TJ III, Zentella A, et al: Shock and tissue injury induced by recombinant human cachectin. Science. 1986; 234: 470-474. [ Links ]
6. Maury CPJ: Tumor necrosis factor-an overview. Acta Med Scand. 1986; 220: 387-394. [ Links ]
7. Scudire P, Lam KS, Ryan KJ, et al: Raised serum levels of tumor necrosis factor in parasitic infections. Lancet. 1986; 11: 1364-1365 [ Links ]
8. Tracey KJ, Beutler B, Lowry SF, et al: Shock and tissue injury induced by recombinant human cachectin. Science. 1986; 234: 470-474 [ Links ]
9. Le J, Vilcek J: Tumor necrosis factor and interleukin-1 cytokines with multiple overlapping biological activities. Lab invest. 1987: 56: 234- 248. [ Links ]
10. Mathison JC, Wolfson E, Ulevith RJ: Participation of tumor necrosis factor in the mediation of gram negative bacterial lipopolysaccharide-induced injury in rabbits. J Clin Invest. 1988; 81: 1925-1937. [ Links ]
11. Seckinger P, Isaaz S, Dayer JM: a human inhibitor of tumor necrosis factor-alpha. J Exp Med. 1988; 167: 1511-1516. [ Links ]
12. Peetre C, Thysell H, Grubb A, Olson I: A tumor necrosis factor binding protein is present in human biological fluids. Exp Hematol. 1988; 14: 414-419. [ Links ]
13. Matsumori A, Yamada T, Susuki H, et al: increased circulating citokines in patients with myocarditid and cardiomiapathy. Hypertension. 1989;13: 706-711 [ Links ]
14. Gulick T, Chung MK, Pieper SJ, et al. interleukin 1 and tumor necrosis factor inhibit cardiac myocite beta-adrenergic responsivness. Proc Natl Acad Sci USA. 1989; 86: 6753-6757. [ Links ]
15. Natanson C, Eichenholz PW, Danner RL, et al: Endotoxin and tumor necrosis factor challenges in dogs simulate profile of human septic shock. J Exp Med. 1989; 169: 823-832. [ Links ]
16. Hosenpud JD, Campbell SM, Mendelson DJ. Interleukin-1-induced myocardial depression in an isolated heart preparation. J Heart Transplant. 1989; 8: 460-464. [ Links ]
17. Ferrari R, AnsndI: Neurohumoral changes in untrated heart failure. Cardiovas Drug Ther. 1989; 3: 979-986. [ Links ]
18. Cavero PG, Miller WL, Heublein, et al: Endothelin in experimental congestive heart failure in the anesthetized dog, am J Physiol. 1990; 259: F312-317. [ Links ]
19. Levine B, Kalman J, Mayer L, et al: Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990; 223: 236-241. [ Links ]
20. Arai K, Lee F, Miyajima A, et al: Citokines: coordinators of immune and inflammatory responses. Ann Rev Biochem. 1990; 59: 783-831. [ Links ]
21. Pye M, Rae AP, Cobbe SM: Study of C-reactive protein concentration in heart failure. Br Heart J 1990; 63: 228-230. [ Links ]
22. McMueeay J, Abdullah I, Dargie HJ, Shapiro D: Increased concentrations of tumor necrosis factor in "cachectic" patients with severe chronic heart failure. Br Heart J, 1991; 66: 356-358. [ Links ]
23. Sampaio EP, Sarno EN, Galilly R, et al: Thalidomide selectively inhibits tumor necrosis factor-alpha production by stimulate human monocytes. J Exp Med. 1991; 173: 699-703. [ Links ]
24. Remes J, Tikkanen I, Fyhrquist, et al: Neuroendocrine activity in untreated heart failure. Br Heart J. 199; 65: 249-255 [ Links ]
25. Haber HL, Leavy JA, Kessler PD, et al: the ESR in congestive heart failure. N Engl J Med. 1991; 324: 353-358. [ Links ]
26. Pagani FD, Baker IS, His C, et al: Left ventricular systolic and diastolic dysfunction after infusion of tumor necrosis factor-alpha in conscious dogs. J Clin Invest. 1992; 90: 389-398. [ Links ]
27. Stevenson LW, Fonarow GC; endothelin and the vascular choir in heart failure. J Am Coll Cardiol. 1992; 20: 854-857. [ Links ]
28. Eichorn FJ: the paradox of beta-adrenergic blockade for the management of congestive heart failure. Am J Med. 1992; 92: 527-538. [ Links ]
29. Dutka DP, Elborn JS, Delarmre F, et al: Tumor necrosis factor-alpha in severe cardiac failure. Br Heart J. 1993; 70: 141- 143. [ Links ]
30. Drexler H, Hayoz D, Munzel T, et al : Endothelial function in congestive heart failure. Am Heart J. 1993; 126: 761-764. [ Links ]
31. Pfeffer MA: Angiotensin-converting enzyme inhibition in congestive heart failure: Benefif and perspective. Am Heart J. 1993; 126; 789-793. [ Links ]
32. Treasure CB, Alexander RW: The dysfunctional endothelium in heart failure. J Am Coll Cardiol. 1993; 22: 129A-134A. [ Links ]
33. Lüscher TF, Noll G: Endothelium-despendent vasomotion in aging, hypertension and heart failure. Circulation. 1993; 87(Suppl. 7): 97-103 [ Links ]
34. Francis GS, Cohn JN, Johnson G, et al: Plasma norepinefhrine, plasma renin activity and congestive heart failure. Relations to survival and the effects of therapy in V- HeFT II. The V-VeFT VA Cooperative Studies Group. Circulation. 1993; 87(Suppl. 6): V140-V148. [ Links ]
35. Yokahoma T, Vaca L, Rossen RD, et al: Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993; 92: 2303-2312. [ Links ]
36. Wiedermann CJ, Bienpold H, Herold M, et al: Increased levels of serum neopterin and decreased production of neutrophil superoxide anions in chronic heart failure with elevated levels of tumor necrosis factor-alpha. J Am Coll Cardiol. 1993; 22: 1897-1901. [ Links ]
37. Ross R: Pathogenesis of atherosclerosis : a perspective for the 19990s. Nature. 1993; 362: 801-809.
38. Zabel P, Schade FU, Schalaak M: Inhibition of endogenous TNF formation by pentoxifylline. Immunobiology. 1993; 187; 447-463. [ Links ]
39. Drexler H: Endothelial dysfunction in heart and potential for reversal by ACE inhibition. Br Heart J. 1994; J72: S11-S14. [ Links ]
40. Hirooka Y, Imaizumi T, Tagawa T, et al: Effects of L-Arginine on impaired acetyl-choline-induced and ischemic vasomotion of the forearm in patients with heart failure. Circulation. 1994; 90: 658-668. [ Links ]
41. Mann DL, Young JB: Basic Mechanisms in congestive heart failure: Recognizing the role of pro inflammatory cytokines. Chest 1994; 105: 897-904. [ Links ]
42. Matsumori A, Yamada T, Susuki H, et al: Increased circulating cytokines in patients with myocarditid and cardiomyopathy. Br Heart J 1194; 72: 651-566. [ Links ]
43. Tsutamoto T, Wada A, Maeda Y, et al: relation between endothelin-1 spillover in the lugs and pulmonary vascular resistance in patients with chronic Herat failure. J Am Coll Cardiol 1194; 23: 1427-1433. [ Links ]
44. Cody RJ; The clinical potential of renin inhibitors and angiotensina antagonists. Drugs 1994; 47: 585-598. [ Links ]
45. Nakamura M, Funakoshi T, Arakawa N, et al: Effect of angiotensina-converting enzyme inhibitors on endothelium-dependent peripheral vasodilatation in patients with chronic heart failure. J Am Coll Cardiol 1994; 24: 1321-1327. [ Links ]
46. Paulus JW: Endothelial control of vascular and myocardial function in heart failure. Cardiovas Drugs Ther. 1994; 8: 437-446. [ Links ]
47. Van der Waal AC, Becker EA, Loos CM, Das PK: Site of rupture orverosion of thrombosescoronary atherosclerotic plaques is characterizedby an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1194; 89: 34-44. [ Links ]
48. Alexander RW: Inflammations and coronary artery disease. N Engl J Med. 1194; 331: 468-469. [ Links ]
49. Kapadia S, Lee JR, Torre-Amione G, et al: Tumor necrosis factor gene and protein expression in adult feline myocardium after endotoxina administration. J Clin Invest. 1995; 96: 1042-1052. [ Links ]
50. Thaik CM, Calderone A. Takahashi N, Colucci WS. Interleukin-Ib modulates the growth and phenotype of neonatal rat cardiac myocites. J Clin Invest. 1995; 96: 1093-1099. [ Links ]
51. Suffredini AF, Reda D, Banks SM, et al: Effects of recombinant dimeric TNF receptors on human inflammatory responses following intravenous endotoxina administration. J Immunol. 1995; 155: 5038-5045. [ Links ]
52. Torre-Amione G, Kapadia S, Lee J, et al: Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995; 92: 1487-1493. [ Links ]
53. Ferrari R, Bachetti T, Confortini R, et al: Tumor necrosis factor soluble receptors in patients with various degrees of congestive heart failure. Circulation. 1995; 92: 1479-1486. [ Links ]
54. Seta Y, Shan K, Bozkurt B, et al: Basic mechanisms in heart failure: the cytokine hypothesis. J Cardiac Fail. 1996; 2: 243-249. [ Links ]
55. Krown KA, Page MT, Nguyen C; et al: Tumor necrosis factor-alpha-induced apoptosis in cardiac myocites; ¿involvement of the sphingolipid signalling cascade in cardiac cell death? J Clin Invest. 1996; 96: 2854-2865. [ Links ]
56. Torre Amione G , Kapadia S, Benedict CR, et al. pro-inflammatory citokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfuncion (SOLVD). J Am Coll Cardiol. 1996; 27: 1201-1206. [ Links ]
57. Torre-Amione G, Kapadia S, Lee J, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human Herat, Circulation. 1996; 93: 704-711. [ Links ]
58. Vanhoutte, PM: Endothelium-dependent Responses in Condestive Heart Failure. J Mol Cell Cardiol. 1996; 28: 2233-2240. [ Links ]
59. Coats AJS: The " Muscle Hypothesis" of Chronic Heart Failure. J Mol Cell Cardiol. 1996; 28: 2255-2262. [ Links ]
60. Kubota T, McTiernan CF, Frye CS, et al: Dilated cardiomyopathy in transgenic mice with cardiac specific overexpression of tumor necrosis factor-alpha. Circu Res. 1997; 81: 627-635. [ Links ]
61. MacGowan GA, Mann DL, Kormos RL et al: Circulating interleukin-6 in severe heart failure. Am J Cardiol. 1997; 79: 1128-1131. [ Links ]
62. Oral H, Dorn GW II, Mann DL: Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian cardiac myocyte. J Biol Chem. 1997; 272: 4836-4842. [ Links ]
63. Yokoyama T, Nakano M, Bednarczyk JL, et al: Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocites. Circulation. 1997; 95: 1247-1252. [ Links ]
64. Eriksson SV, Kjeshus J, Eneroth P, Swedberg K. Neopterin, tumor necrosis factor, C-reactive protein and prostaglandin E2 in patients with severe congestive heart failure treated with enalapril. Circulation. 1997; 96(suppl I): I-322. Abstract. [ Links ]
65. Kapadia S, Oral H, Lee J, et al: Hemodyamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ Res. 1997; 81: 187-195. [ Links ]
66. Olivetti G, Abbi R, Quaini F, et al: Apoptosis in the failing human heart. N Engl J Med. 1997; 336: 1131-1141. [ Links ]
67. Anker SD, Chua TP, Ponikowski P, et al: Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation. 1997; 96: 526-534. [ Links ]
68. Ridker PM, Cushman M, Stampfer MJ, et al: Inflammation, aspirine, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997; 336: 973-979. [ Links ]
69. Anker SD, Egerer KR, Volk HD, et al: Elevated Soluble CD14 receptors and altered cytokines in chronic heart failure: Am J Cardiol. 1997; 79: 1426-1429. [ Links ]
70. Bryant D, Becker L, Richardson J, et al: Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation. 1198; 97: 1375-1381. [ Links ]
71. Hetts SW: To die or not to die. An overview of apoptosis and its role in desease. JAMA. 1998; 279: 300-307. [ Links ]
72. Bozkurt B, Kibbs S, Clubb Jr M, et al: Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular disfunction and remodelling in rats. Circulation. 1998; 97: 1382-1391. [ Links ]
73. Panas D, Khadour FH, Szabo C, Schultz R. Proinflammatory cytokines depress cardiac afficiency by a nitric oxide-dependent mechanism. Am J Physiol. 1998; 275: H 1016-H 1023. [ Links ]
74. Meldrum DR: Tumor necrosis factor in the heart. Am J Physiol. 1998; 274: R 577-R 595. [ Links ]
75. Kapadia S, Dibbs Z, Kerrelmeyer K, et al: The role of cytokines in the failing human heart. In: Crawford M, ed. Cardiology Clinics. Philadelphia, Pa: WB Saunders; 1998: 645- 656. [ Links ]
76. Tsutamoto T, Hisanaga T, Wada A, et al: Interleukin-6 spillver in the peripheral circulation increases with the severity of heart failure, and high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol. 1998; 31: 391-398. [ Links ]
77. Sliwa K, Skudicky D, Candy G, et al: Randomized investigation of effects of penttoxifylline on left ventricular performance in idiopathic dilated cardiomyopathy. Lancet. 1998; 351: 1091-1093. [ Links ]
78. Ceconi C, Curello S, Banchetti T, et al: Tumor necrosis factor in congestive heart failure: a mechanism of disease for the new millennium?. Prog Cardiovas Dis. 1998; 41 (suppl I): 25-30. [ Links ]
79. Hasper D, Hummel M, Kleber FX, et al: Systemic inflammation in patients with heart failure. Eu Heart J. 1998; 19: 761-765. [ Links ]
80. Givertz MM, Colucci WS: New targets for heart-failure therapy: endothelin, inflammatory cytokines, and oxidative stress. Lancet. 1998; 352 (suppl I) SI34-SI38. [ Links ]
81. Koller-Strametz J, Pacer R, Frey B, et al: Circulating tumor necrosis factor-alpha levels in chronic Herat failure: relation to its soluble receptor II, interleukin-6, and neurohumoral variables. J Heart Lung Transplant. 1198; 17: 356-362. [ Links ]
82. Franco F, Thomas GD, Giroir BP, et al: Magnetic resonance imaging and invasive evaluation of development of heart failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation. 1999; 99: 448-454. [ Links ]
83. Kumar A, Brar R, Wang P, et al: Role of nitric oxide and cGMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol. 1999; 276: R265-R276. [ Links ]
84. Agnoletti L, Curello S, Banchetti T, et al: Serum from patients with severe heart failure dowregulates eNOS and is proapoptotic: role of tumor necrosis factor-alpha. Circulation; 1999; 100: 1983-1991. [ Links ]
85. Mann DL: Mechanisms and models in heart failure: a combinatorial approach. Circulation; 1999; 100; 999-1088. [ Links ]
86. Gullestad L, Aukrust P, Ueland T, et al: Effect of high-versus low-dose angiotensina converting enzyme inhibition on cytokine levels in chronic failure. J Am Coll Cardiol. 1999; 34: 2061-2067. [ Links ]
87. Dibss Z, Thornby J, white BG, Mann DL: Natural variability of circulating levels of cytokines and citokine receptors in patients with heart failure: implications for clinical trials. J Am Coll Cardiol. 1999; 33: 1935-1942. [ Links ]
88. Niebauer J, Volk HD, Kemp M,et al: Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999; 353: 1838-1842. [ Links ]
89. Deswal A, Bozkurt B, Seta Y, et al: A phase I trial of tumor necrosis factor receptor (p75) fusion protein (TNFR:Fc) in patients with advanced heart failure. Circulation. 1999; 99: 3224-3226. [ Links ]
90. Ross R: Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340: 115-126. [ Links ]
91. Liu L, Zhao SP: The changes of circulating tumor necrosis factor levels in patients with congestive heart failure influenced by therapy. Int J Cardiol. 1999; 69: 77-82. [ Links ]
92. Anker SD, Rauchhaus M: Insights into the pathogenesis of chronic heart failure: immune activation and cachexia. Curr Opin Cardiol. 1999; 14: 211-216. [ Links ]
93. Herrera-Garza E, Cubillos-Garzón A, Stetson, SJ, et al: Factor de necrosis tumoral-alfa: un mediador en la patogénesis de la insuficiencia cardíaca. Arch Inst Cardiol Mex. 1999; 69: 462-468. [ Links ]
94. Opie LH: ACE Inhibitors for Congestive Heart Failure. En Opie LH. Angiotensin Converting Enzyme Inhibitors. The Advance Continues. University of Cape Town Press. Third Edition, 1999; 6: 131-153. [ Links ]
95. Koenig W, Sund M, Froelich M, et al: C-reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy men: results from the MONICA study (Monitoring Trends and Determinants in Cardiovascular Diesease). Ausburg Cohort Study, 1984 to 1992. Circulation. 1999; 99: 237-242. [ Links ]
96. Prabhu SD, Chandrasekar B, Murray DR, Freeman GI: Beta-adrenergic blockade in developing heart failure: effect on myocardial inflammatory cytokines, nitric oxide, and remodelling. Circulation. 2000; 101: 2103-2109. [ Links ]
97. Baumgarten G, Knuefermann P, Mann DL: Cytokines as emerging targets in the treatment of heart failure. Trends Cardiovasc Med. 2000; 10: 216-223. [ Links ]
98. Bozkurt B, Chee A, Lee-Jackson D, et al: Results of an open label dose response study of thalidomide in patients with advanced failure and elevated levels of TNF. J Am Coll Cardiol. 2000; 35: 172A. Astract. [ Links ]
99. Rauchhaus M, Doehner W, Francis DP: Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000; 102: 3060-3067. [ Links ]
100. Kalra D, Baumgarten G, Dibbs Z, et al: Nitric oxide provokes tumor necrosis factor-alpha expression in adult feline myocardium through a cGMO-dependent pathway. Circulation. 2000; 102: 1302-1307. [ Links ]
101. Mann DL, Knueferman P, Baumgarten G: Cytokines in ischemic heart disease. Dialogues in Cardiovascular Medicine. 2000; 5: 135-146. [ Links ]
102. Agnoletti, L, Comini, L: Do Cytokines and endothelial function have an impact on myocardial activity. Dialogues in Cardiovascular Medicine. 2000; 5: 149-153. [ Links ]
103. Drexler H, Schieffer B: What are the implications of the interaction between the neurohomoral system and the cytokines? Dialogues in Cardiovascular Medicine. 2000; 5: 154-161 [ Links ]
104. Anker SD: Has the time arrived to use anticytokine therapy in chronic heart failulre? Dialogues in Cardiovascular Medicine. 2000; 5:162-170. [ Links ]
105. Ridker PM, Hennekens C H, Buring J E, Rifai N: C-reactive protein and others marker of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000; 342: 836-843. [ Links ]
106. Sharma R, Coats AJS, Anker SD: The role of inflammatory mediators in chronic heart failure: Cytokines, nitric oxide, and endothelin-I. Int J Cardiol. 2000; 72: 175-186. [ Links ]
107. Bolger A P, Anker SD: Tumor necrosis factor in chronic heart failure: a peripheral view on pathogenesis, clinical manisfestations and therapeutic implications. Drugs. 2000; 60: 1247-1257. [ Links ]
108. Pomerantz BJ, Reznikow IL, Harken AH, Dinarello CA: inhibition of capase I reduces human myocardial ischemic dysfunction via inhibition of II-18 and II-I beta. Proc Natl Acad Sci USA. 2001; 98: 2871-2879. [ Links ]
109. Lisman KA, Stetson SJ, Koerner MM, et al: Managing heart failure with immunomodulatory agents. En: Crowford M. ed. Cardioly Clinics./Philadelphia, Pa: W. B. Saunders, 2001: 617-625. [ Links ]
110. Sivasubramanian N, Coker ML, Kurrelmeyer KM, et al: Left ventricular remodelling in transgenic mice with cardiac restricted over expression of tumor necrosis factor. Circulation. 2001; 104: 826-831. [ Links ]
111. Fischtscherer S, Rossig L, Breuer S, et al: Tumor necrosis factor antagonism with etanercept improves systemic endothelial vasorreactivity patients with advanced heart failure. Circulation. 2001; 104: 3023-3025. [ Links ]
112. Wei GC, Sirois M G, Qu R, et al: Effects of quinapril on myocardial function, ventricular remodelling, and cardiac cytokine espression in congestive heart failure in the rat. Cardiovasc Drugs The. 2001;14: 234- 235. [ Links ]
113. Gurlek A, Kilickap M, Dincer I, et al: Effect of losartan on circulating TNF-alpha levels and left ventricular systolic performance in patients with heart failure. J Cardiovasc Risk. 2001; 8: 279-282. [ Links ]
114. Gullestad L, Ueland T, Brunsvig A, et al: Effect of metoprolol on cytokine levels in chronic heart failure: a substudy in the metoprolol Controlled-Release Randomised Intervention Trial in Heart Failure (MERIT-HF). Am Heart J. 2001; 141: 418-421. [ Links ]
115. Deswal A, Misra A, Bozkurt B: The role of anti-cytokine therapy in the failing heart. Heart Fail Rev. 2001; 6: 143-151. [ Links ]
116. Bozkurt B, Torre-Amione G, Warren MS, et al: Results on the targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. 2001; 103: 1044-1047. [ Links ]
117. Mann DL. Recents instgts into the role of tumor necrosis factor in the failing heart, Heart Fail Rev. 2001; 6: 71-80. [ Links ]
118. Krum H: The Theraoeutic Role of Beta-adrenoreceptor-Blocking Agents. En Eydén L. Prevention of Disease Progression Throughout the Cardiovascular Continuum. The Role of Adrenergic Blockade. Springer-Verlag Berlin Heidelberg New York. 2001; 96-106. [ Links ]
119. Mann DL: Inflammatory Mediators and the Failing Heart. Past, Pressent, and the Foreseeable Future. Circ. Res. 2002; 91: 988-998. [ Links ]
120. Anker SD, Coats AJS: How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol. 2002; 86: 123-130 [ Links ]
121. Albert CM, Rifai N, Stampfer MJ, Ridker PM: Prospective study of C-Reactive Protein, homocysteine, and plasma lipid level as predictors of sudden cardiac death. Circulation. 2002; 105: 2595-2599. [ Links ]
122. Pradhan AD, Manson JE, Rossuw JE, et al: Inflammatory biomarkers, hormone replacements therapy, and incident coronary heart disease: prospective analysis from the omen´s Health Initiative observational estudy. JAMA. 2002; 288: 980-987. [ Links ]
123. Libby P, Ridker PM, Masseri A: Inflammation and atherosclerosis. Circulation. 2002;105:1135-1143. [ Links ]
124. Buffon A, Biasucci IM, Liuzzo G, et al: Widespread coronary inflammation in unstable angina. N Engl J Med. 2002; 347: 5-12. [ Links ]
125. Ridker Pm: On Evolutionary Biology, Inflammation, Infection, and the Causes of Atherosclerosis. Circulation. 2002; 105: 2-4 [ Links ]
126. Mann DL: Comunicación Personal. 2003, Enero 23.I [ Links ]
* Profesor de Medicina,Facultad de Medicina,Universidad de El Salvador,
** Fellow, Cardiología Intervencionista, Baystate Medical Center – Tufts University (West Campus), Massachusetts, U.S.A.
*** Profesor de Medicina,Facultad de Ciencias de la Salud; Universidad Salvadoreña "Alberto Masferrer " El Salvador..

