










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Pediátrica Costarricense
versión impresa ISSN 1409-0090
Acta pediátr. costarric vol.21 no.1 San José ene. 2009
Original
Expresión fenotípica en pacientes con fibrosis quística
(Phenotypic expression in patients with cystic fibrosis)
Carolina Murillo Guevara1,
José Pablo Gutiérrez Schwanhauser2
1. Posgrado en Pediatría, Sistema de Estudios de Posgrado, Universidad de Costa Rica.
Correspondencia: Dra. Carolina Murillo Guevara, correo electrónico: gmurillo@racsa.co.cr
2. Servicio de Neumología, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera", Caja Costarricense de Seguro Social
Resumen
Objetivo: Como nuestro país carece de un análisis descriptivo y a largo plazo de los pacientes con Fibrosis Quística, nuestro objetivo principal en esta investigación, es determinar la expresión fenotípica y la variación en la mortalidad en los pacientes con diagnóstico de FQ en el Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera", en los 20 años comprendidos entre 1983 y 2003.
Métodos: Es un estudio descriptivo, con información recopilada en forma retrospectiva, basado en los registros y expedientes médicos de los pacientes, los casos fueron identificados a través del registro electrónico del Servicio de Neumología.
Resultados: En el período comprendido entre el primero de enero 1983 y el 31 de diciembre 2003 se diagnosticaron 59 casos con Fibrosis Quística, que cumplieron los criterios de inclusión ( expresión fenotípica de la enfermedad y valores de cloruros en sudor igual o mayor a 60 mEq/L).
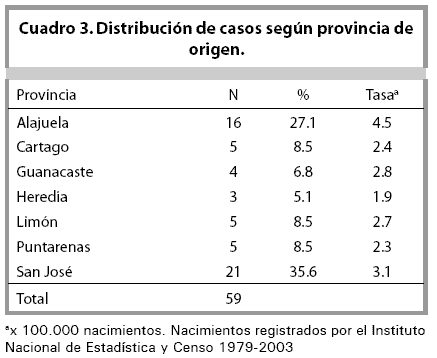
Las provincias con mayor frecuencia de casos fueron Alajuela y San José.
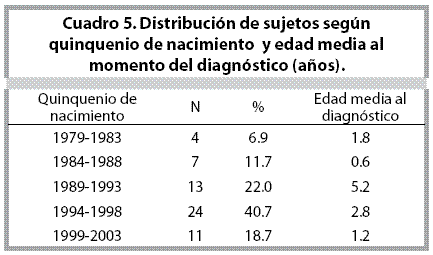
La mayoría de estos niños fueron diagnosticados antes de los 3 años (n=44, 74.6%); la edad media al momento del diagnóstico fue de 2 años 7 meses.
En cuanto a la frecuencia de síntomas y signos al momento del diagnóstico, la presentación más frecuente fueron los síntomas pulmonares en 37 pacientes (62.7%). Cuatro sujetos tenían historia familiar positiva de Fibrosis Quística.
Se registró un promedio de 6.2 hospitalizaciones por paciente; y el rango varió de 0 a 31 internamientos por sujeto.
Entre las complicaciones se identificaron: prolapso rectal, colelitiasis, hepatopatía, cirrosis hepática, hemoptisis, Diabetes Mellitus, hipocratismo digital.
En el análisis de los cultivos de esputo, se encontró más frecuentemente Pseudomona aeruginosa (n= 30).
De los 59 pacientes, 14 fallecieron (23.7%). La edad media al momento del fallecimiento fue de 5 años 9 meses. Se reportó una media de sobrevida de 156.8 meses y una mediana de 141 meses.
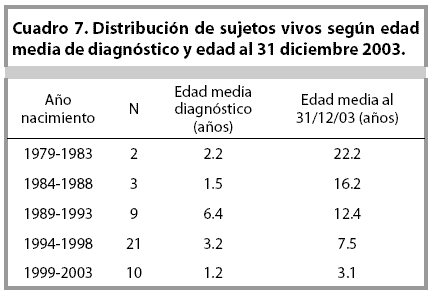
De los 45 pacientes que permanecían vivos para el 31 de diciembre del 2003, la edad media fue de 8 años 9 meses. La paciente con mayor edad fue de 24 años 4 meses.
Conclusiones: En general, los pacientes con Fibrosis Quística viven más que en el pasado, pero todavía presentan infecciones pulmonares crónicas y otras complicaciones médicas relacionadas a su enfermedad, incluyendo diabetes, bronconeumonía, hepatopatía con cirrosis, prolapso rectal y hemoptisis.
Descriptores: fibrosis quística, fenotipo, epidemiología, población pediátrica, complicaciones, bacteriología, diagnóstico.
La Fibrosis Quística (F)Q es una enfermedad pulmonar genéticamente determinada que afecta principalmente a la raza caucásica1 y representa una causa importante de morbilidad pulmonar y gastrointestinal en niños.2 Su incidencia varía desde 1 de cada 3000 nacidos vivos de raza blanca hasta 1 de cada 90000 de Asiáticos3, y varía basado en la distribución étnica en diferentes regiones. Debido a las diferencias en la composición étnica latinoamericana, se debe esperar menor incidencia de FQ en la población de Centro y Suramérica que en otros caucásicos1; lo cual es un indicador del diferente comportamiento de la enfermedad de acuerdo a la población en que se presenta.
La primera descripción entendible clínica y patológica de FQ se acredita a Anderson en 1938.4
Durante los primeros años de la década de los 80 se identificó el defecto fisiológico fundamental (falla en la regulación del AMPc en el transporte de cloruro). En 1985, el defecto, heredado en un patrón autosómico recesivo, se localizó en el cromosoma 7. En 1989, el gen de FQ se identificó por clonación.5
Este nuevo conocimiento ha sugerido nuevas terapias; se ha aprendido más acerca de la enfermedad pulmonar y se han abierto nuevas líneas de investigación básica.
La FQ sigue diagnosticándose básicamente por clínica. En la glándula sudorípara, el transporte electrolítico anormal por su conducto genera sudor que tiene una concentración anormal de sodio y cloruro, por ello el diagnóstico se basa en una concentración de cloruro en sudor mayor a 60 mEq/L.
La enfermedad pulmonar es la principal causa de morbilidad y es responsable del 95% de la mortalidad. En el pulmón, las vías aéreas son el sitio inicial de compromiso, con aclaramiento mucociliar anormal, infecciones a repetición, y bronquiectasias que llevan a una disfunción pulmonar progresiva y falla respiratoria. En el páncreas, la obstrucción de los conductos lleva a atrofia y a la necesidad de reemplazo enzimático pancreático en aproximadamente el 85% de los pacientes. En algunos pacientes, la destrucción del páncreas también lleva a insuficiencia endocrina pancreática y a Diabetes Mellitus. En el tracto genital masculino, la obstrucción del vas deferens ocasiona esterilidad en más del 95% de los hombres. En el hígado, este tipo de secreciones dentro de los conductos biliares conlleva a cirrosis biliar focal en algunos pacientes. En el intestino, la deshidratación del contenido intestinal provoca íleo meconial en aproximadamente 5-15% de los recién nacidos4. Además, la enfermedad se asocia muy frecuentemente con alteración en el crecimiento. Su presentación varía en diferentes edades, y la severidad de la enfermedad y su tasa de progresión en los órganos involucrados varían considerablemente. Aunque la mayoría de los pacientes tienen insuficiencia pancreática y necesitan terapia de reemplazo de enzimas exógenas, aproximadamente el 15% tienen suficiente función pancreática exocrina residual para permitir la digestión normal sin suplementos enzimáticos.6 El término "suficiencia pancreática" se ha utilizado para describir la condición en un grupo de pacientes, generalmente de mayor edad al diagnóstico y con niveles inferiores de cloruros en sudor, enfermedad respiratoria leve, crecimiento normal y mejor pronóstico que los pacientes con insuficiencia pancreática.
Mundialmente se han analizado en los últimos años las implicaciones clínicas de las diferentes mutaciones. La mutación ΔF508 estα presente en, al menos, el 70% de los cromosomas de FQ. Se sabe que la frecuencia de la mutación ΔF508 es considerablemente mayor en países de Europa del Norte como Inglaterra, Francia, Dinamarca y Norteamérica, y la frecuencia es considerablemente menor en países de Europa del Este y del Sur como Italia, Portugal, España y Grecia. Algunas de las otras mutaciones publicadas representan del 1 al 3 % de los cromosomas de FQ y son de particular importancia para el estudio de los portadores. Estas incluyen la G542X, la G551D, la R553X, la W1282X y la N1303K.7
Al menos una parte de la variabilidad en la presentación clínica de FQ se explica por las diferencias en las mutaciones del CFTR (Regulador de conductancia transmembrana de FQ). La función pancréatica, por ejemplo, está muy relacionada con la mutación específica que el paciente porta y las mutaciones asociadas con la función pancreática exocrina adecuada son dominantes sobre aquellas asociadas con insuficiencia pancréatica. Las mutaciones asociadas con la función pancréatica eficiente son predominantemente las que tienen actividad de canal detectable (ejemplo, R117H y A455E) , mientras que las mutaciones con poca actividad (ΔF508, G551D) o las que resultan de reducción cuantitativa severa del CFTR (W1282X) se asocian con insuficiencia pancreática.
En la vía aérea, la relación entre genotipo y fenotipo es menos clara, probablemente refleja una mayor influencia del ambiente al que las vías aéreas están expuestas, lo que contribuye a la variabilidad. Sin embargo, algunas mutaciones se asocian con un curso clínico leve de la enfermedad. Los pacientes con función pancreática adecuada tienen un curso pulmonar más leve, que los pacientes con insuficiencia pancreática. En general, las mutaciones que resultan en una actividad reducida pero no ausente del CFTR se asocian con enfermedad más leve.5
En 1993 se publicó en el Journal of Pediatrics8 un artículo analizando el cambio en la epidemiología de la FQ, siendo esta publicación la más cercana a nuestras intenciones. Este estudio reportó los datos de 17857 pacientes con FQ que se incluyeron en 1990 en el registro de la "Fundación de Fibrosis Quística". Se analizaron las características demográficas, tasas de supervivencia, función pulmonar, antropometría, datos microbiológicos, tasas de complicaciones, utilización de centros de salud; y se compararon con datos similares recolectados en 1969, 1972 y 1978. Los hallazgos más notables fueron que la proporción de pacientes adultos aumentó 4 veces entre 1969 y 1990; la edad media al momento del diagnóstico en 1990 para todos los pacientes en el registro de FQ fue 12.5 años; FQ se diagnosticó en 90% de los pacientes a la edad de 12 años; la edad promedio de sobrevida se duplicó entre 1969 y 1990, de 14 a 28 años y el patógeno respiratorio reportado más frecuentemente fue la Pseudomona aeruginosa.
En general, se concluyó, que los pacientes con FQ viven mucho más que en el pasado, pero que todavía tienen infecciones pulmonares crónicas y otras complicaciones médicas relacionadas con su enfermedad, incluyendo diabetes, obstrucción intestinal, cirrosis, hemoptisis, y neumotórax. Los investigadores han empezado a correlacionar las características clínicas, anormalidades fisiológicas, y las mutaciones específicas de FQ.8
La FQ era prácticamente desconocida en Costa Rica; fue a partir de finales de los años setentas que se empezó a pensar en su diagnóstico.
El Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera" (HNN) cuenta con una extensa documentación de pacientes con FQ, sin embargo carece de una base de datos y de un análisis de la misma, lo que hace difícil determinar la realidad de esta enfermedad en nuestro medio. Factores como incidencia, distribución de casos en el país, momento en el cual se hace el diagnóstico, síntomas de presentación más frecuentes, hospitalizaciones, evolución clínica, gérmenes más frecuentemente aislados y mortalidad, así como otros, nos son desconocidos. Es por esto que pretendemos ordenar toda esta información en una base de datos, con el fin de realizar un análisis sistemático y determinar la expresión fenotípica de los pacientes con diagnóstico de FQ en el HNN en los últimos 20 años.
Materiales y métodos
Este es un estudio con propósitos académicos, original, de carácter observacional, descriptivo, retrospectivo basado en registros de laboratorio y expedientes médicos.
El período de estudio incluyó desde el primero de enero de 1983 hasta el 31 de diciembre de 2003 (20 años), y se identificaron los sujetos de acuerdo al registro estadístico del HNN y del Servicio de Neumología. Se incluyeron a todos los pacientes con prueba de cloruros en sudor positiva10 (niveles superiores o iguales a 60 mEq/L) y con manifestaciones clínicas de la enfermedad. Se excluyeron a los pacientes con prueba de cloruros en sudor positiva pero sin expresión fenotípica, así como los niños con expresión fenotípica pero con prueba de cloruros en sudor negativa.
Para cada caso se recopilaron los datos clínicos y socio-demográficos mediante la revisión de los respectivos expedientes clínicos. Los datos fueron obtenidos sistemáticamente mediante la utilización de una hoja de recolección de las variables de interés , luego se introdujeron los datos en el programa estadístico Epi info9, y posteriormente se realizó la determinación de las frecuencias de cada una de las variables para obtener el perfil fenotípico para cada paciente.
Para el análisis de los datos se realizó la determinación de las medidas de tendencia central en las variables cuantitativas y la determinación de frecuencias y proporciones en las variables cualitativas. Todas las determinaciones fueron realizadas por medio del software estadístico Epi Info 3.4.3 (CDC-2007).
El protocolo fue presentado al Comité Etico- Científico de la Unidad de Investigación del HNN y recibió la aprobación (CLOBI-HNN-005-2005).
Resultados
En el período comprendido entre el primero de enero de 1983 al 31 de diciembre de 2003, se identificaron a 59 pacientes que cumplieron con los criterios de inclusión para este estudio.
En el cuadro 1 se describen las características de la población estudiada.
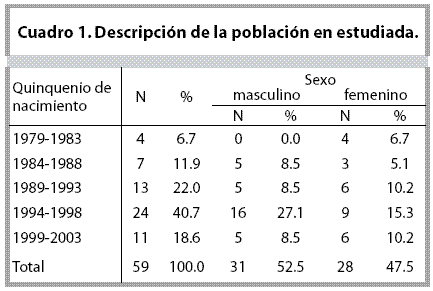
Durante el quinquenio de 1994-1998 se presentó la tasa de frecuencia más elevada, seguido por 1989-1993 y 1999-2003 (Cuadro 2).

El cuadro 3 muestra la distribución por provincia de origen de los casos estudiados. La provincia de Alajuela tuvo una tasa de incidencia de 4.5 casos por cada 100.000 nacimientos y San José 3.1 casos por cada 100.000 nacimientos.
El 74.6% de los casos fueron diagnosticados antes de los tres años (N= 44) y de estos, el 52.3% antes del año de edad; el rango es de 18 días a 11 años. La edad media al momento del diagnóstico fue de 2.1 años (Cuadros 4 y 5).

El 69.5% de estos niños presentaron más de un síntoma o signo asociado al momento del diagnóstico. Las manifestaciones pulmonares como tos crónica, sibilancias a repetición , neumopatía crónica, bronconeumonía y atelectasia se presentó en 37 pacientes (62.7%), seguido de falla para progresar, pobre ganancia de peso o pérdida de peso en 20 de ellos (33.8%); los síntomas gastrointestinales como enfermedad diarreica crónica, dolor abdominal, vómitos, deposiciones fétidas se presentaron en 17 pacientes (28.8%), en 6 la presentación inicial fue de ileo meconial, en 3 sujetos se encontró hepatomegalia al momento del diagnóstico, así como prolapso rectal en 2 , pancreatitis en 1 y otitis recurrente en otro niño. En 4 casos el diagnóstico se facilitó por tener historia familiar de FQ (Cuadro 6).

El 35.6% (N= 21) de los sujetos presentaron complicaciones: 3 prolapso rectal, 2 colelitiasis, 6 hepatopatía, y 3 de estos cirrosis hepática, 3 hemoptisis, 3 Diabetes Mellitus tipo 1, y 2 hipocratismo digital. Además, cuatro pacientes requirieron colocación de gastrostomía y uno transplante pulmonar bilateral.
Al analizar el cultivo de esputo se encontró Pseudomona aeruginosa en 30 sujetos, seguido de Streptococo alfa hemolítico en 17, Staphylococo aureus en 14 pacientes y Streptococo pyogenes, Klebsiella pneumoniae, Staphylococo coagulasa negativo en 8 pacientes. Otros gérmenes encontrados fueron Haemophilus influenzae, Acinetobacter sp, Neisseria sp, Escherichia coli, Enterobacter sp, Stenotrophomona maltophila, Serratia marcescens, Streptococo pneumoniae, Candida albicans y Proteus sp. En 21 pacientes no se aisló germen. La edad media de colonización con Pseudomona aeruginosa es de 4.4 años (16.6% para menores de un año y 86.6% para 8 años).
Se registró un promedio de 6.2 hospitalizaciones por niño, con un rango desde 0 hasta 31 internamientos por sujeto durante los 20 años de estudio. Catorce (23.7%) pacientes fallecieron. La edad media al momento del fallecimiento fue de 5.7 años (1 mes – 12.3 años ).
De los 45 pacientes que permanecían vivos para el 31 de diciembre del 2003, 24 eran hombres y 21 mujeres, con una edad media para esa fecha de 11.3 años . La paciente con mayor edad fue de 24.3 años (Cuadro 7).
Se reportó una media de sobrevida de 156.8 meses (IC 95% 123.8-189.7) y una mediana (tiempo donde el 50% de los pacientes han fallecido) de 141 meses (Figura 1).
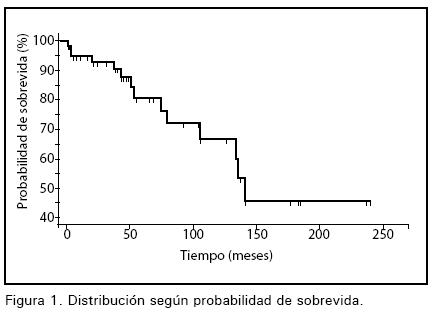
Discusión
En los 59 pacientes que se incluyeron en este estudio no hubo diferencia ignificativa entre el sexo masculino y femenino; y se evidenció que las provincias más afectadas fueron Alajuela y San José.
Con respecto a la tasa de incidencia, se evidenció que la tasa más elevada se presentó durante el quinquenio de 1994-1998, seguido por 1989-1993 y 1999-2003.
El mayor porcentaje de pacientes diagnosticados con FQ nacieron posterior al año 1996; no está claro el por qué. Entre las posibles razones podemos mencionar que en los últimos años se ha dado más educación y conocimiento sobre la enfermedad, así mismo, se dio la consolidación de la Clínica de FQ en el HNN. Se puede observar también, la relación de más sujetos diagnosticados a partir del año 1994.
La mayoría fueron diagnosticados antes de los tres años de edad, y de estos, la mayoría antes del año de edad. La edad media del diagnóstico para esta revisión fue de 2.08 años. Si comparamos estos datos con otras publicaciones en países desarrollados, por ejemplo, en el estudio de Farell y colaboradores (Wisconsin, Estados Unidos), la edad media del diagnóstico, separando a los niños diagnosticados por tamizaje, fue de 95.8 semanas (un año). Por el contrario el diagnóstico por tamizaje se realizó tan tempranamente como 12.4 semanas11. Tomando en cuenta estos datos, y que en nuestro medio el diagnóstico actual continúa realizándose básicamente por sospecha clínica, se podría considerar que se hace relativamente tardío. Shwachman y colaboradores12 crearon la definición actual de diagnóstico temprano de FQ dentro de los primeros 3 meses de edad. Ya se ha demostrado que los pacientes diagnosticados por presentación clínica después de los 2 meses de edad manifiestan los peores resultados clínicos a pesar de recibir niveles más altos de terapia a largo plazo por al menos 10 años13. Por el contrario si comparamos nuestros resultados con los obtenidos en América Latina14, por ejemplo, en el artículo publicado en 1991 posterior a un seguimiento de 10 años en 4 países latinoamericanos (Brazil, México, Chile y Argentina) en donde la edad media de diagnóstico fue mayor a 3 años, se podría considerar que nuestros datos son mejores que estos. El diagnóstico temprano a través de tamizaje neonatal es posible después 1979 cuando Crossley y colaboradores determinaron la medición de tripsinógeno inmunoreactivo para identificar niños con FQ11. El tamizaje para FQ provee una oportunidad de maximizar el potencial clínico de los pacientes cuya supervivencia, de otra manera sería limitada a la adultez temprana. Entre sus ventajas se incluyen lo oportunidad de reorganizar el cuidado y enfatizar estrategias proactivas preventivas como la eliminación de la mal nutrición y potencialmente la erradicación de P. aeruginosa cuando aparece por primera vez11.
La presentación clínica más frecuente fueron las manifestaciones pulmonares, seguido de falla para progresar y síntomas gastrointestinales. El 50% de los pacientes con FQ presentan en la infancia compromiso respiratorio, falla para progresar o ambos15. El tracto respiratorio casi siempre está involucrado en FQ, y las complicaciones pulmonares usualmente dominan el cuadro clínico3.
Se aisló más frecuentemente Pseudomona aeruginosa en el cultivo de esputo de estos niños, obteniéndose una edad media de colonización de 4.4 años. En el estudio de Farell y colaboradores11, la edad media de colonización por este germen fue de 6.04 años. Esto demuestra que en nuestro medio la colonización es más temprana. Cada vez es más claro que hay factores de riesgo potenciales para la infección por P. aeruginosa en niños con FQ; estudios incriminan antibióticos orales por largo tiempo, uso aerosoles, desnutrición, exposición persona-persona, y características intrínsecas como insuficiencia pancreática. Aunque algunos de estos factores son fundamentalmente iatrogénicos, se debe reconocer que al tiempo del diagnóstico en el Centro de FQ de los Estados Unidos, cerca de un tercio de los pacientes con FQ registrados ya han adquirido P. aeruginosa y la adquisición/infección ocurre en la infancia temprana12. Los efectos por P. aeruginosa se conocen desde hace muchos años, pero el impacto cuantitativo de este patógeno en niños en relación a morbilidad y mortalidad se ha vuelto más evidente durante la década pasada. Por ejemplo, Emerson y colaboradores16 recientemente reportaron que los pacientes positivos por P. aeruginosa con FQ tienen 2.6 veces más riesgo de muerte. Hudson y colaboradores17 encontraron mayor mortalidad si la P. aeruginosa fue detectada en cultivos antes de los 2 años de edad.
La mayoría de los fallecidos (N= 6) nacieron en los años 1979- 1988. Entre 1996-2002 se realizó un diagnóstico más temprano de la enfermedad pero la edad media de fallecimiento también fue más temprana, esto en relación a que el paciente que falleció al mes de edad por ileo meconial correspondió a este grupo.
Sólo la mitad de los sujetos que nacieron entre 1979-1983 permanecían vivos para el final del estudio, a pesar de que la edad media del diagnóstico fue de 2.2 años; podría ser a causa de menor abordaje integral ya que no se había consolidado la Clínica de FQ al igual que menos posibilidades terapéuticas. El 76.3% del total de los niños permanecían vivos para el 31 de diciembre de 2003; si lo comparamos con el estudio Latinoamericano18, en donde casi la mitad permanecía vivo en el período de seguimiento y su edad media fue baja (6.4 años en Brazil, 7.4 años en Chile, 9.6 en México, y 11.3 en Argentina) demostramos que nuestros resultados son superiores, ya que no sólo es mejor el porcentaje de niños vivos, sino, que la edad media es superior (11.3 años) en un período de estudio similar.
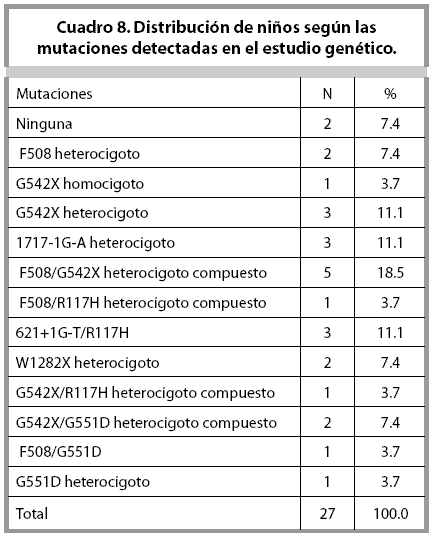
El laboratorio de Genética Molecular del HNN identifica desde el año 2005, 10 mutaciones14: ΔF508, G542X, G551D, ΔI507, R560T, W1282X, N1301K, R117H, 1717-1G-A, 621+1G-T, por tanto, pacientes que presentan otras mutaciones aparte de estas asociadas no pudieron ser identificados.
Es de suma importancia implementar el tamizaje neonatal para FQ en Costa Rica, ya que priorizará los métodos de cuidado para estos niños ya que los distintos resultados han confirmado que el diagnóstico temprano per se no asegura un mejor pronóstico, pero provee una buena oportunidad .
Abreviaturas: FQ, fibrosis quística; HNN, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera".
Referencias
1. Macri C, Gentile A, Manterola A. Epidemiology of cystic fibrosis in Latin America. Pediatr Pulmonol. 1991. 10: 249- 253 [ Links ]
2. Behrman, Kliegman, Arvin. Nelson Tratado de Pediatría. 15 ed. California: MacGraw-Hill Interamericana. 1997. [ Links ]
3. McMillan, DeAngelis, Feigin, Warshaw. Oski´s Pediatrics Principles and Practice. 3era ed. Baltimore: Lippincott. Williams and Wilkins. 1999. [ Links ]
4. Welsh M. The path of discovery in understanding the biology of Cystic Fibrosis and approaches to therapy. Am J Gastroenterol. 1994; 89: S97-S105. [ Links ]
5. Davis P.B. Cystic Fibrosis. Am J Respir Crit Care Med. 1996;154: 1229-56. [ Links ]
6. Kerem E, Corey M, Kerem, Bat-Sheva A. The relation between genotype and phenotype in Cystic Fibrosis- Analysis of the most common mutation (ΔF508). N Engl J Med 1990; 323: 1517-22. [ Links ]
7. Beaudet A. Genetic testing for cystic fibrosis. Pediatr Clin North Am 1992 ; 39: 213-226. [ Links ]
8. FitzSimmons S. The changing epidemiology of cystic fibrosis. J Pediatr 1993; 122: 1-9. [ Links ]
9. Bar-Zohar D, Segal-Algranati D, Belson A, et al. Diagnosing cystic fibrosis— asthma and failure to thrive as indications for sweat test. J Med 2004; 35: 93-103. [ Links ]
10. Farrell PM, Li Z, Kosorok MR, et al. Bronchopulmonary disease in children with cystic fibrosis after early or delayed diagnosis. Am J Resp Crit Care Med 2003; 168: 1100-8. [ Links ]
11. Shwachman H, Redmond A, Khaw KT. Studies in cystic fibrosis: report of 130 patients diagnosed under 3 months of age over a 20-year period. Pediatrics 1970; 46: 335-343. [ Links ]
12. Sims EJ, Clark A, McCormick J, et al. Cystic fibrosis diagnosed after 2 months of age leads to worse outcomes and requires more therapy. Pediatrics 2007; 119: 19-28. [ Links ]
13. Ferrie RM, Schwarz MJ, Robertson NH. Development, multiplexing, and application of ARMS tests for common mutations in the CFTR gene. Am J Hum Genet 1992; 51: 251-262. [ Links ]
14. Hay WW, Hayward AR, Levin MJ, Sondheimer JM. Current Pediatric Diagnosis and Treatment. 15th ed. Denver: McGraw Hill; 2001. [ Links ]
15. Emerson J, Rosenfeld M, Mc Namara S, et al. Pseudomona aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 2002; 34: 91-100. [ Links ]
16. Hudson VL, Wielinski CL, Regelmann WE. Prognostic implications of initial oropharyngeal bacterial flora in patients with cystic fibrosis diagnosed before the age of two years. J Pediatr 1993; 122: 854-860 [ Links ]
17. Macri CN, de Gentile AS, Manterola A, et al. Epidemiology of cystic fibrosis in Latin America: preliminary communication. Pediatr Pulmonol 1991;10: 249-53. [ Links ]
18. Anglani F, Camporese C, Picci L. Cystic fibrosis genetics in Southern Europe. Lancet 1990; 336: 379-380. [ Links ]
