










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Pediátrica Costarricense
Print version ISSN 1409-0090
Acta pediátr. costarric vol.19 n.2 San José Jan. 2005
Diagnóstico molecular de un caso de Deficiencia de Acil CoA Deshidrogenasa de Cadena Media (MCADD) detectado por MS/MS.
Andrey Sequeira1,
Abelino Sánchez 2,
Carlos Vargas 3,
Alejandra Reuben 4,
Julio Rivera 5,
Rafael Trejas6,
Carlos de Céspedes 7,
Manuel Saborío 8.
1. Bach en Biología, Biólogo molecular, Laboratorio de biología molecular del Centro para la Prevención de Discapacidades, Hospital Nacional de Niños. Correspondencia: Centro para la Prevención de Discapacidades, frente a entrada de Emergencias del Hospital Nacional de Niños, tel. (506) 258-2818, ext. 2405, fax (506) 258-2516, bioaseq@yahoo.com, asequeirac@tamizajeneonatal.com
2.Lic. en Microbiología. Jefe Laboratorio de espectrometría de masas en tandem (MS/MS) del Centro para la Prevención de Discapacidades, Hospital Nacional de Niños.
3. Lic. en Medicina y Cirugía, especialista en Pediatría, médico Pediatra, Servicio de genética y metabolismo, Hospital Nacional de Niños.
4. Lic. en Microbiología. Laboratorio de espectrometría de masas en tandem (MS/MS) del Centro para la Prevención de Discapacidades, Hospital Nacional de Niños.
5. Lic. en Microbiología. Jefe Citogenética y Biología Molecular, Hospital Nacional de Niños.
6. Lic. en Microbiología. Director Laboratorio para el Tarnizaje Neonatal, Centro para la Prevención de Discapacidades, Hospital Nacional de Niños.
7. PhD en Bioquímica. médico asesor, Servicio de genética y metabolismo, Hospital Nacional de Niños.
8. Lic. en Medicina y Cirugía, especialista en Pediatría, especialista en Genética médica. Médico Pediatra, Servicio de genética y metabolismo, Director Programa Nacional de Tamizaje, Centro para la Prevención de Discapacidades, Hospital Nacional de Niños.
Resumen
La Deficiencia de Acil CoA Deshidrogenasa de Ácidos Grasos de Cadena Media (MCADD; OMIM 201450) es el trastorno de la β-oxidación más comúnmente diagnosticado en humanos. Se hereda de forma autosómica recesiva y presenta una frecuencia que varía entre 1:4900 y 1:25000. La mutación más frecuente es la A985G que se observa en condición homocigoto en 81% de los casos y heterocigoto compuesto en 18%. El bloqueo enzimático lleva a la acumulación de ésteres de acilcarnitinas de cadena media lo cual permite la detección temprana de la enfermedad mediante el análisis de Espectrometría de Masas (MS/MS) y su posterior confirmación molecular.
El primer caso de MCADD en Costa Rica fue detectado por MS/MS en el Laboratorio Nacional de Tamizaje y la presencia de la mutación A985G fue confirmada por métodos moleculares. La mutación afecta una región lejana del sitio activo que participa en el empaquetamiento de la proteína, proceso que es asistido por los chaperones moleculares. Hasta la fecha, el paciente permanece asintomático lo ucal podría explicarse por una serie de factores genéticos y ambientales que permiten mantener, parcialmente, la actividad de la enzima.
Palabras claves: tamizaje neonatal, acilcarnitinas, mutación, empaquetamiento, MCADD.
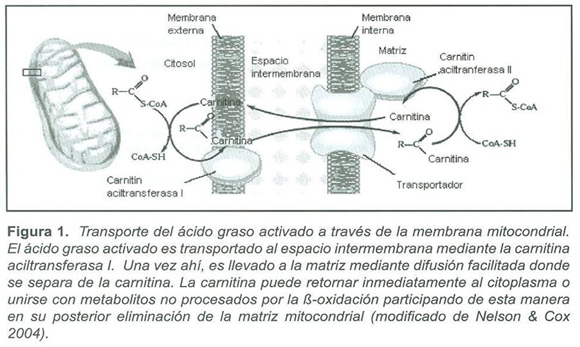
Durante los años 70`s se describieron trece trastornos hereditarios de la oxidación de ácidos grasos (FAO), causados por la deficiencia de transportadores y enzimas de membrana, o de enzimas de la matriz mitocondrial, donde se lleva a cabo la β-Oxidaciσn (1) (fig. 1)
La Deficiencia de Acil CoA deshidrogenasa de Ácidos Grasos de Cadena Media (MCADD; OMIM 201450) es el trastorno de la β-oxidación de ácidos grasos más comúnmente diagnosticado en humanos(2,3). El defecto se hereda de forma autosómica recesiva y la prevalencia de esta enfermedad es alta en poblaciones de origen caucásico, principalmente en aquellas que descienden de los asentamientos del norte de Europa. La frecuencia estimada por programas de tamizaje neonatal que utilizan Espectrometría de Masas en Tandem (MS/MS) varía en un rango que va de 1:4 900 a 1: 25 000, lo cual está determinado por la estructura genética de cada población estudiada(4). Se considera que la deficiencia es poco común en poblaciones hispanas. Esta posición se ha visto cuestionada; sin embargo, por resultados obtenidos en un estudio piloto a través de análisis de espectrometría de masas en California, EUA, se requiere mayor investigación al respecto.
Los pacientes con MCADD lucen sanos al nacimiento, la enfermedad se presenta generalmente entre los 3 y los 24 meses de edad, aunque se han reportado casos confirmados que se mantienen asintomáticos hasta la adultez(5). Los síntomas pueden ser hipoglicemia, vómito, letargo, encefalopatía, paro respiratorio, hepatomegalia, apnea, paro cardiaco, coma y muerte súbita. A largo plazo, los pacientes pueden presentar problemas de desarrollo sicomotor y comportamiento, debilidad muscular crónica, falla para progresar, parálisis cerebral y trastorno de déficit atencional(6,7). Muchos de estos síntomas se presentan durante el aumento en la actividad física y/o el ayuno prolongado.
El gen ACADM (OMIM 607008) se localiza en la región 31 del brazo corto del cromosoma 1 (1p31), está formado por 12 exones distribuidos a lo largo de 44 kilobases y codifica un monómero precursor de 421 aminoácidos(8). La proteína MCAD madura es una de las cuatro deshidrogenasas de Acil CoA dependientes de la longitud de la cadena que inician la β-oxidación de ésteres de Acil CoA(9). Estas cuatro enzimas catalizan la oxidación de ácidos grasos de cadena corta (4-6 carbonos), media (6-10 carbonos), larga y muy larga (12-18 carbonos), para producir un doble enlace entre los átomos de carbono 2 y 3 como resultado de la deshidrogenación. Son enzimas solubles localizadas en la matriz mitocondrial con excepción de la acil CoA deshidrogenasa de cadena muy larga que se encuentra unida a la membrana interna mitocondrial(10,11). El empacamiento del péptido en la mitocondria es asistido por las chaperonas moleculares.
La mutación puntual A985G en el gen MCAD, que causa el cambio de una lisina por un glutamato en la posición 304 (K304E) en la proteína madura, se presenta en el 90% de los alelos de pacientes con la deficiencia. De los pacientes clínicamente diagnosticados con MCADD, 81% son homocigotos para A985G y un 18% son heterocigotos compuestos para esta mutación(2,4,6-7,9,12).
La frecuencia de portadores oscila entre 1:40 a 1:100(4). Es posible que no todos los pacientes homocigotos para A985G muestren niveles elevados de octanoilcarnitina (C8) en el periodo neonatal (ver adelante). Posiblemente otros polimorfismos presentes en las chaperoninas podrían ofrecer cierta protección al estabilizar la proteína mutante y normalizar la actividad enzimática. Las otras mutaciones en el heterocigoto compuesto son raras y diversas. Se han descrito 45 mutaciones hasta el momento aunque la relación genotipo-fenotipo no ha sido bien determinada siendo A985G la más estudiada(2,4,13).
El proceso de la ß-oxidación en la matriz mitocondrial es precedido por la asociación de carnitina con ácidos grasos activados (acilCoA), lo que permite su transporte a través de la membrana mitocondrial interna(14) (Fig.1). Se ha determinado que pacientes con trastornos hereditarios del metabolismo de ácidos grasos, eliminan en la orina acilcarnitinas específicas que reflejan la acumulación anormal de tioésteres de acil CoA. Esto permite que la determinación de tales concentraciones, mediante técnicas como MS/MS, se convierta en una herramienta diagnóstica invaluable para la detección y posterior confirmación por métodos moleculares de errores de ß-oxidación causantes de diferentes enfermedades(15).
El incremento de ésteres de acil CoA de cadena media producidos por el bloqueo enzimático lleva a un incremento de ácidos grasos de cadena media, ácidos dicarboxílicos, acilglicinas y acilcarnitinas de cadena media en plasma y orina (Fig. 1, 2). La carnitina se une a residuos acil y participa en su eliminación (Fig. 1), disminuyendo la concentración de residuos conjugados con CoA e incrementando la relación entre CoA libre y acilado.
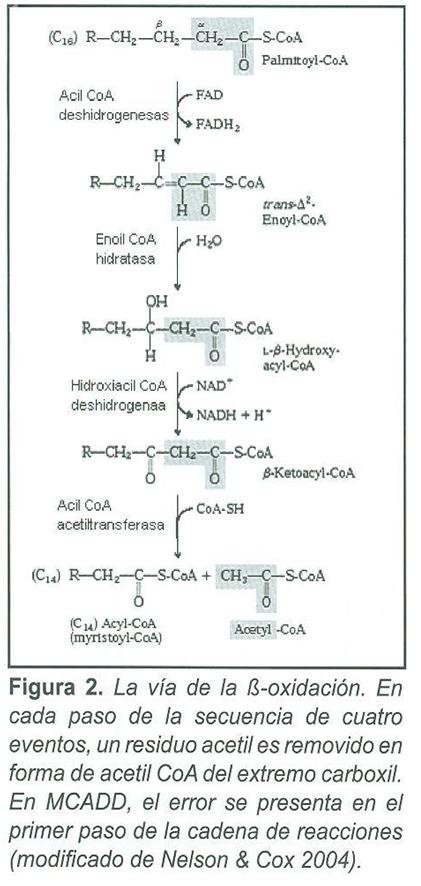
Pacientes con MCADD presentan un incremento de octanoilcarnitina (C8 >0.23 µmol/L) y un aumento de la relación octanoilcarnitina-decanoilcarnitina (C8/C10 >5) (16). De esta manera, el análisis por MS/MS permite, en principio, el diagnóstico de todos los pacientes con la deficiencia de MCAD sin importar la mutación, estado sintomático o tratamiento.
El presente reporte expone un perfil amplio del análisis y diagnóstico del primer caso de MCADD detectado en Costa Rica dentro del Programa Nacional de Tamizaje Neonatal mediante espectrometría de masas en tandem (MS/MS) y confirmado a través de procedimientos moleculares(17).
Materiales y métodos:
El análisis de espectrometría de masas en tandem y el análisis de ADN se llevó a cabo en los laboratorios de espectrometría de masas y biología molecular respectivamente, del Centro para la Prevención de Discapacidades del Hospital Nacional de Niños "Dr. Carlos Sáez Herrera".
Espectrometría de Masas en Tandem (MS/MS): la muestra fue recolectada por tamizaje masivo en papel de filtro S&S 903 al 4° día de edad y analizada con las siguientes condiciones: Sistema MRM, para todos los ésteres de acilcarnitinas a saber C2, C3, C4, C5, C6, C8, C10, C12, C14, 016 y sus derivados, con una voltaje del cono de 35 Volt, Energía de colisión de 27 Volt, flujo de inyección de 10 uI/min, un flujo del gas desolvatante (N2) de 450 lit/Min, utilizando una mezcla de acetonitrilo 80:20 con agua, tiempo de escaneo en el detector de 1.50 minutos.
Extracción de ADN y análisis molecular: muestras de sangre fueron recolectadas en tubos con K3EDTA tanto del paciente como de la madre con el fin de determinar sus genotipos con respecto a la mutación A985G. La extracción de ADN se realizó siguiendo el protocolo basado en NaCI previamente publicado(18). Con respecto al análisis molecular, la mutación A985G en el exón 11 del gen MCAD introduce un sitio de restricción Ncol en el mutante y no en el alelo normal.
De esta manera, el alelo mutante puede ser distinguido del normal mediante una digestión para Ncol del producto de la Reacción en Cadena de la Polimerasa (PCR) seguida por la separación de productos en un gel de agarosa(19,12). El ADN correspondiente del probando y de la madre fue amplificado mediante la PCR utilizando los siguientes imprimadores: sentido, 5’-TTTATGCTGGCTGA-AATGGCCATG; antisentido, 5’-AGTTTTTCTACAGGATAT TCTGTATTAAATCCATGGCCTC. El producto fue digerido con la enzima Ncol (Fermentas).
Caso clínico:
El paciente fue referido por resultado anormal de tamizaje neonatal masivo: Octanoyl-carnitina (C8) en 2.36µM (punto de corte: 0.23µM) y Hexanoyl-carnitina (punto de corte : 027µM). Fue evaluado por primera vez en la Consulta de Enfermedades Metabólicas del Hospital Nacional de Niños a los 15 días de edad. Madre de 16 años de edad, G1,P1,A0, con control prenatal, sin enfermedades asociadas a la gestación. Antropometría al nacer: Peso: 3540 gramos, Talla: 51 cm, CC: 33 cm. Apgar: 9-9. No hay antecedentes patológicos familiares de relevancia. No hay historia de retraso mental, hipoglicemias o muerte súbita. Consanguinidad en tercer grado (el padre del niño es hermano del abuelo por línea materna).
En su evaluación inicial:, Gases venosos normales, glicemia en 91 mg/dl, con cetonemia negativa. Transaminasa oxalacética discretamente elevada en 99 lU/ L pruebas metabólicas en orina: normales. Hemograma normal. US de hígado normal. Exploración cardiovascular que incluye Ecocardiografía normal. Evaluación de control a los 21/2 meses con evidencia de elevación de transaminasas hepáticas, con TSGP: 164 IU/L, TSGO: 254 IU/ L, Fosfatasa alcalina: 174 IU/L, Glicemia: 94 mg/dI, Cetonemia: negativa. Con electrolitos séricos y pruebas de función renal normales. A sus 5 meses de edad, TSGO: 225 IU/L, TSGP: 163 IU/L, Cetonemia trazas, con glicemia: 98 mg/dl, bilirrubinas totales: 0.4 mg/ dl, B. Directa: 0.1 mg/ dl. Las serologías virales por citomegaIovirus, hepatitis, VDRL son negativas. Tiene un examen físico normal, con desarrollo psicomotor y antropométrico adecuado y totalmente asintomático hasta la fecha de preparación de este informe. (16 de agosto del 2005).
El análisis de mutaciones para MCADD mostró una condición homocigota para la mutación A985G.
Desde el punto de vista terapéutico, el manejo ha sido nutricional, con tomas de leche materna frecuentes, evitando el ayuno. Al iniciar la ablactación se indicó dieta rica en carbohidratos complejos alternando con 5 tomas de leche al día. Se instruyó a la madre para que evite el ayuno y se mantenga vigilante ante las enfermedades propias de la infancia que podrían ser desencadenantes de crisis.
Resultados y discusión
La implementación técnica de metodologías de MS/MS y biología molecular permite el tamizaje metabólico y la confirmación diagnóstica de gran variedad de trastornos en neonatos, por ejemplo, enfermedades metabólicas hereditarias del metabolismo de los aminoácidos (enfermedad del Jarabe de Arce, Fenilcetonuria, Homocistinuria, etc.) y, al incorporar el análisis de perfiles de acilcarnitinas, transtornos relacionados con la oxidación de ácidos grasos y otros ácidos orgánicos(20,21). El Programa de Tamizaje Neonatal de Costa Rica analiza, anualmente, las muestras de 72.000 neonatos a través de diferentes tecnologías dentro de las cuales MS/MS y biología molecular juegan un papel fundamental en el proceso de detección y confirmación diagnóstica.
El análisis molecular de la muestra confirma la presencia de la mutación A985G en condición homocigota. El paciente presenta la deficiencia de MCAD aunque, al momento de la presentación de este artículo, se mantenía asintomático. La variación afecta el empaquetamiento y la estabilidad de la enzima. El proceso de empaquetamiento es monitoreado por sistemas de control de calidad, los cuales incluyen las chaperonas moleculares que asisten el empaquetamiento, y proteasas intracelulares, que eliminan proteínas mal empaquetadas.
Puesto que la eficiencia de estos sistemas depende de las condiciones celulares, por ejemplo, de temperatura y nivel energético, y probablemente de diferencias genéticas asociadas entre individuos, el efecto de esta y otras variaciones en la secuencia de ADN con pérdida de sentido no puede, en general, ser predicho. Recientemente se ha demostrado que este tipo de mutaciones, además de influenciar la biogénesis de la proteína, también afecta la eficiencia del editaje de intrones o "splicing" al interferir con los sitios de unión para factores moduladores del editaje. Este fenómeno se ha observado en algunas deshidrogenasas y está en proceso de ser caracterizado para los genes MCAD y VLCAD(8). La sintomatología variable que puede presentar la enfermedad podría explicarse por el hecho de que el efecto que la mutación pueda tener, in vivo, podría estar modulado por factores celulares y genéticos.
Investigaciones sobre la toxicidad de ácidos grasos de cadena media, por ejemplo el ácido octanoico, muestran propiedades narcóticas que pueden contribuir, de manera significativa, al letargo y coma hepático observado en pacientes durante períodos de descompensación metabólica. Con respecto a la deficiencia de energía, la manifestación no tan severa es probablemente causada por una combinación de, primero, el hecho de que varios ciclos de oxidación pueden proceder antes de que la vía sea bloqueada y, segundo, alguna actividad enzimática puede surgir de la acil CoA deshidrogenasa de cadena larga (LCAD) y SCAD debido a que existe traslape en cuanto a los sustratos en los que actúan (8,11).
Desde el punto de vista de la patogénesis molecular, la mutación A985G, que lleva al reemplazo de la lisina por el ácido glutámico, se localiza lejos del centro activo. La sustitución K304E afecta el empaquetamiento de la proteína MCAD normal llevando a la deficiencia en la expresión de ésta. Específicamente, la proteína MCAD es asistida en su empaquetamiento intramitocondrial, primero, por la chaperona de choque térmico 70 (Hsp7O) y, subsecuentemente, por la chaperonina Hsp60, proceso que se ve afectado en presencia de la sustitución. La lisina en la posición 304 es trascendental para el empaquetamiento eficiente del monómero ya que la interacción de cargas entre la lisina 304 y el aspartato 346 es determinante para el ensamblaje del tetrámero y, por lo tanto, para la estabilidad de la enzima(8). Esto tiene efecto directo en la conformación del sitio activo y en el surco de unión al sustrato, lo cual podría contribuir a la patogénesis de la variante.
Aproximadamente el 20% de los pacientes muere antes de ser diagnosticados y un porcentaje importante de los sobrevivientes al primer episodio de descompensación aguda presentan secuelas significativas(22). Es de gran importancia mencionar que las deficiencias en el metabolismo de ácidos grasos han sido extensamente estudiadas en casos donde se presenta el síndrome de muerte súbita y de estos, la mutación A985G es la mejor estudiada. No obstante, menos del uno por ciento de los casos de muerte súbita investigados presentan esta mutación. Esto aunado al hecho de que se han observado perfiles bioquímicos característicos para trastornos de la oxidación de ácidos grasos en un número considerable de casos indica la importancia de la investigación de otros padecimientos diferentes al MCADD(23).
El diagnóstico molecular de MCADD es atractivo porque se ha observado que, en general, el 80% de los casos son homocigotos para la mutación A985G. Sin embargo, resultados tempranos de algunos estudios indican una frecuencia de homocigotos mucho menor de lo esperado para la mutación y una frecuencia mayor de heterocigotos compuestos, esto podría tener implicaciones importantes en cuanto a expresión fenotipica y diagnóstico molecular(4 y 7).
Hay una variación importante en la severidad de diferentes mutaciones, la actividad enzimática residual generada por diversas mutaciones puede ser modulada por factores endógenos, tales como el número de chaperonas disponible y la temperatura. La acción de genes modificadores podría tener un efecto importante en la variación del fenotipo producido. Mutaciones que directamente inhabilitan el sitio activo pueden tener consecuencias más críticas que aquellas que afectan los sitios de editaje, el marco de lectura, el empaquetamiento o la estabilidad. A pesar de la heterogeneidad de factores involucrados, la severidad intrínseca de las mutaciones se ve reflejada en el fenotipo bioquímico y se expresa como con diferentes perfiles de acilcarnitina(2). El estudio molecular de aquellos que exhiban concentraciones elevadas de octanoilcarnitina permitirá estimar la prevalencia y determinar la frecuencia de la mutación A985G en la población costarricense. Si bien las restantes mutaciones no podrán ser identificadas, la información obtenida permitirá llevar a cabo una evaluación precisa sobre la eficiencia de la técnica para el diagnóstico general de la deficiencia de MCAD, además de un mayor conocimiento de la estructura genética de nuestra población.
Por otro lado, el diagnóstico de esta y otras enfermedades a través de tales tecnologías (MS/ MS y biología molecular), es de importancia trascendental para el tratamiento adecuado de estos casos. Como se mencionó anteriormente, el paciente se mantiene asintomático. No obstante, se puede aplicar tratamiento médico preventivo (uso de L-carnitina) así como la vigilancia de factores que, dada la etiología del padecimiento, podrían precipitar la manifestación clínica, como lo son el ayuno prolongado y el aumento de la actividad física o infecciones. Ambas medidas disminuyen considerablemente la probabilidad de manifestación y, en caso de presentarse, la severidad de los síntomas.
Referencias
1. Karpati G, Carpenter S, Engel AG, et al. The syndrome of systemic carnitine deficiency: clinical, morphologic, biochemical, and pathophysiologic features. Neurology 1975; 25:16-24. [ Links ]
2. Andresen BS, Dobrowolski SF, O’Reilly L et al. Medium-chain acyl-coA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001 ; 68:1408-18. [ Links ]
3. Comeau AM, Larson C, Eaton R. Integration of new genetic diseases into Statewide Newborn Screening: New England Experience. Am J Med Genet 2004; 125C: 35-41. [ Links ]
4. Maier EM, Liebl B, Roschinger W et al. Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-coA dehydrogenase deficiency. Hum Mutat. 2005; 25:443-52. [ Links ]
5. Feillet F, Steinmann G, Vianey-Saban C et al. Adult presentation of MCAD deficiency revealed by coma and severe arrythmias. Intensive care Med 2003; 29:1594-97. [ Links ]
6. Wang SS, Fernhoff P, Hannon WH. Medium chain acyl-CoA dehydrogenase human genome epidemiology review. Genet Med 1999; 1:332-9. [ Links ]
7. Carpenter K, Willey V, Sim KG et al. Evaluation of newborn screening for medium chain acylCoA dehydrogenase deficiency in 275 000 babies. Arch Dis Child Fetal Neonatal Ed. 2001; 85:F105-F9. [ Links ]
8. Gregersen N, Bross P. Andresen B. Genetic defects in fatty acid ß-Oxidation and acyl-CoA dehydrogenases: molecular pathogenesis and genotype-phenotype relationships. Eur J Biochem 2004; 271: 470-82. [ Links ]
9. Andresen B, Bross P, Udvari S et al. The molecular basis of medium acyl-CoaA dehydrogenase (MCAD) deficiency in compound heterocigous patients: is there a correlation between genotype and phenotype. Hum Mol Gen 1997; 6: 695-707. [ Links ]
10. Kim P, Miura R. Acyl- CoA dehydrogenases and Acyl-CoA oxidases: structural basis of mechanistic similarities and differences. Eur J Biochem 2004; 271: 483-93. [ Links ]
11. Bartlett K, Eaton S. Mitochondrial ß-Oxidación. Eur J Biochem 2004; 271:462-69. [ Links ]
12. Clayton P, Doig M, Ghafari S et al. Screening for medium chain acyl-coA dehydrogenase deficiency using electrospray ionisation tandem mass spectrometry. Arch Dis Chiid 1998; 79:109—15. [ Links ]
13. Tajima G, Sakura N, Ono H et al. Medium-chain acyl-coA dehydrogenase deficiency found through newborn screening by tandem mass spectrometry in Japan. J Inherit Metab Dis 27 2004; (Suppl 1:11-20. [ Links ]
14. Nelson DL, Cox MM. Lenhinger: Principles of biochemistry. 4th ed, Worth Publishers. United States of America. 2004: 598-619. [ Links ]
15. Schulze A, Linder M. Kohlmüller D et al. Expanded newborn screening for inborn errors of metabolism by electrospray ionization- tandem mass spectrometry: Results, outcome and implications. Pediatrics 2003; 111:1399-1406. [ Links ]
16. Chace DH. Hillman SL, Van Hove JL et al. Rapid diagnosis of MCAD deficiency: quantitatively analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin Chem 1997; 43:2106-13. [ Links ]
17. De Céspedes C, Saborio M, Trejos R et al. Evolution and innovations of the National Neonatal and High Risk Screening Program in Costa Rica. Revista de Biología Tropical 2004; 52:451-66. [ Links ]
18. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research 1988.16: 1215-1224. [ Links ]
19. Yokota I, Indo Y, Coates PM et al. Molecular basis of medium chain acyl-coenzyme A dehydrogenase deficiency: an A-to-G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J. Clin. lnvest. 1990; 86: 1000-3. [ Links ]
20. Magera MJ, Lacey JM, Casetta B, Rinaldo P. Method for the determination of total homocysteine in plasma by stabie isotope dilution and electrospray tandem mass spectrometry. Clin Chem 1999;45:1517-22. [ Links ]
21. Burtis CA, Ashwood ER, Tietz NW. Using tandem mass spectrometry for metabolic disease screening among newborns: a report of a work group. Morb. Mortal. Wkly. Rep. 2001; 50 1917-28 [ Links ]
22. Schoen E, Baker JC, Colby C et al. Cost-Benefit Analysis of Universal Tandem Mass Spectrometry for Newborn Screening. Pediatrics 2002; 110: 781-6. [ Links ]
23. Opdal S, T Rubdum. The Sudden lnfant Death Syndrome gene: does it exist? Pediatrics 2004; 114:506-12. [ Links ]
