








Serviços Personalizados
Journal
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkActa Pediátrica Costarricense
versão impressa ISSN 1409-0090
Acta pediátr. costarric vol.18 no.1 San José Jan. 2004
Tumor de Wilms con ambigüedad genital: revisión y reporte de un caso
Julieta Solís 1, Carlos Rodríguez 2, Luis Corrales 3, Roberth Moya 4
1 Médico Pediatra.
2 Pedíatra Asistente, Servicio de Oncología, Hospital Nacional de Niños.
3 Médico General.
4 Médico Residente de Pediatría.
Dirección para correspondencia. Correo electrónico: jusolis@hotmail.com, Apartado postal: 2065-3000.
Acta Pediátrica Costarricense 2004; volumen 18, número 1.
En este articulo se presenta un caso de Tumor de Wilms diagnosticado con ambigüedad genital, el cual ingresó al servicio de Oncología del Hospital Nacional de Niños con una historia de un aumento del volumen abdominal progresivo. El paciente fue estudiado y se le diagnosticó un tumor de Wilms en riñón izquierdo. El tumor de Wilms es asociado en algunas ocasiones a ciertas patologías como hemihipertrofias, aniridias, y ambigüedad genital. Se debe tener presente que al encontrarse cualquiera de estas alteraciones se debe estudiar por un tumor de Wilms.
Palabras claves: Tumor de Wilms, ambigüedad genital, masa renal, tumor renal.
Caso
Se trata de un paciente masculino que al año y 2 meses consultó por un cuadro caracterizado un aumento del volumen abdominal progresivo de 4 días de evolución. La madre refería también que cuando el niño caminaba o comía iniciaba con dificultad respiratoria que cedía espontáneamente. El niño no asocia fiebre, pérdida de peso o apetito. vómitos o ningún otro síntoma. Como único antecedente personal patológico es una ambigüedad genital por lo que está en control. En el examen físico de ingreso se describe un abdomen con una masa dura que ocupa casi todo el hemiabdomen izquierdo, no dolorosa de aproximadamente 12 cm por 10 cm. También se detalla un hermafroditismo con hipospadias.
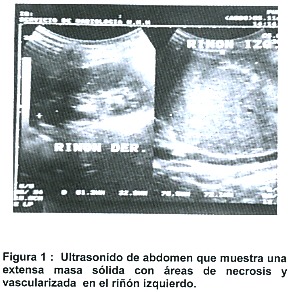
Al día siguiente de su ingreso se le realiza un ultrasonido de abdomen en donde se reporta una extensa masa sólida que mide 12.7 cm por 10,3 cm por 7,2 cm con áreas de necrosis y vascularizada en el riñón izquierdo y que atraviesa línea media. (Figura 1)
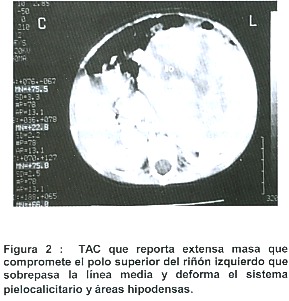
Por tal motivo se le coordina una tomografía axial computarizada que reporta que se observa una extensa masa que compromete el polo superior de riñón izquierdo, de más o menos 12 cm por 10 cm de diámetro y 10 cm de altura, que sobrepasa la línea media y que deforma el sistema pielocalicitario y muestra áreas hipodensas probablemente en relación con áreas de necrosis. Riñón derecho de tamaño y forma normal. (Figura 2)
A la semana posterior a su ingreso, al paciente se le realiza una biopsia, guiada por ultrasonido, de la masa en hemiabdomen izquierdo la cual reporta un Tumor de Wilms, por lo que se le inicia quimioterapia con Vincristina, Actinomicina. Posterior a la segunda dosis de quimioterapia se le realiza un ultrasonido el cual reporta una masa sólida de 12.4 cm por 10.8 cm por 8,6 cm que comparada al ultrasonido anterior se mantiene del mismo tamaño pero con más áreas de necrosis.
El paciente ha continuado en control en Oncología en donde se le ha observado una disminución del perímetro abdominal al pasar de 60 cm previo a la quimioterapia a 48,5 cm en la ultima cita control. Actualmente ha recibido 5 ciclos de quimioterapia.
Discusión
El tumor de Wilms es un enfermedad curable en la mayoría de los casos afectados. Más del 90% sobreviven 4 años después del diagnóstico(1). El pronóstico está relacionado no solo con la etapa de la enfermedad en el momento del diagnóstico, las características histopatológicas del tumor, y el tamaño del tumor, sino también con la estrategia en equipo para cada paciente por parte del cirujano pediatra (1-3) El tumor de Wilms en menos de un 10 % se presenta en niños con malformaciones conocidas. Los fenotipos relacionados con el tumor de Wilms pueden clasificarse en síndromes de sobrecrecimiento o sin sobre crecimiento, el síndrome de sobre crecimiento es el resultado de un excesivo crecimiento somático prenatal y postnatal, lo cual resulta macroglosia, nefromegalia y hemihipertrofia. Los dos tumores de sobrecrecimiento más comunes relacionados con el tumor de Wilms son el síndrome de Beckwith Wiedemann y la hemihipertrofia aislada. (4-8)
Los trastornos de no sobre crecimiento relacionados con el tumor de Wilms incluye la aniridia aislada, la trisomía 18, aniridia combinada con malformaciones genitourinarias y el síndrome de retardo mental (AGR), síndrome de Bloom y el de Denys-Drash (9). El tumor de Wilms (hereditario o esporádico) parece resultar de cambios en uno o más de varios genes. Algunas mutaciones específicas de línea germinal de uno de estos genes localizado en el brazo corto del cromosoma 11 , no sólo están relacionados con el tumor de Wilms sino que también causan una variedad de problemas genitourinarios tales como el criptorquidismo e hipospadias (10), así como el síndrome poco común conocido como Denys-Drash. El tumor de Wilms es asociado en algunas ocasiones con ambigüedad genital, la cual se presenta por la poco virilización de los genitales externos masculinos o excesiva virilización de los femeninos (11). Durante la diferenciación sexual normal, el feto, que es bipotencial, tiene una tendencia innata a desarrollarse como mujer. Sin influencia de una cascada de eventos iniciada tempranamente en la vida del feto, por el factor determinante testicular (SRY), los genitales internos y externos serán femeninos. Así la diferenciación normal masculina requiere de:
1- Una acción adecuada del SRY.
2- Producción testicular de testosterona y de la sustancia inhibidora mulleriana (hormona antimulleriana).
3- Producción normal de gonadrotropinas por el eje hipotálamo hipofisiario ( en el segundo y tercer trimestre de gestación)
4- Conversión de testosterona a dihidrotestosterona (por encima de la 5 alfa reductasa).
5- Respuesta adecuada de órganos blanco a andrógenos.
Tipos
Pseudohermafroditismo Masculino; Individuo 46 XY con genitales extemos ambiguos. Generalmente por:
1. Falla de la corteza suprarrenal en la producción de precursores de hormonas esteroideas.
2. Falla testicular en producir adecuadas de testosterona cantidades
3. Falta de respuesta a andrógenos por parte de órganos blanco
Pseudohermafroditismo Femenino: Individuo 46 XX con amplia gama de genitales externos ambiguos desde cliteromegalia hasta fusión labioescrotal y sinus urogenital. Generalmente asociado a hiperplasia suprarrenal congénita (HSC) o exceso de producción materna de andrógenos (HSC materna, tumores adrenales u ováricos, luteomas del embarazo)
Hermafrodita Verdadero: Independiente del cariotipo, existe tejido testicular (con túbulos seminíferos) y ovarios (con folículos de Graff maduros). El cariotipo más frecuente es 46 XX. La presentación clínica es de ambigüedad genital, pero puede ser un masculino casi normal o femenino casi normal. El componente testicular usualmente es inútil, el componente ovárico puede ser incluso funcional.
Asociación con malignidades y tumores:
La mayoría de los casos de ambigüedad genital asociada a tumores, en especial Tumor de Wilms, son esporádicos (por delección 11 pI3), sin embargo existe una forma familiar causada por una translocación balanceada en los padres. (11)
A pesar del numero de genes que parecen estar implicados en el desarrollo del tumor de Wilms, el tumor hereditario de Wilms no es común, teniendo del 1% a12% de los pacientes una historia positiva familiar de tumor de Wilms. (12-13) El riesgo de padecer el tumor de Wilms entre los descendientes de personas que han tenido tumores unilaterales (o sea, esporádicos) es bastante bajo (< 2%). (14). Los hermanos de niños con Tumor de Wilms tienen una probabilidad baja de desarrollar este tumor (12). Entre el 4% y el 5% de los pacientes tienen tumores de Wilms bilaterales, pero por lo general no son hereditario (12 -13)
Aunque la mayoría de los pacientes con un diagnostico histológico del Tumor de Wilms responden bien a los tratamientos actuales, aproximadamente 10% de los pacientes tienen características histopatológicas que están asociadas con un pronostico mas precario, y, en algunos tipos, con una alta incidencia de recidiva y muerte. El tumor de Wilms puede dividirse en 2 grupos pronósticos sobre la base de su histopatología.
1. Histología favorable - histológicamente imita el desarrollo del riñón normal y consiste de tres componentes: blastema, epitelio, (túbulos) y estroma. No existe anaplasia.
2. Histología no favorable - caracterizada por anaplasia anaplásico (pleomorfismo celular y atipia extremos, difusos) (15). La anaplasia focal puede no conferir un diagnostico tan precario como la anaplasia difusa. La anaplasia esta relacionada con la resistencia a la quimioterapia y podría ser detectada después de aplicar quimioterapia preoperatoria.(16-17)
Sobre el Tumor de Wilms se ha creado un sistema de clasificación clinicopatológico según el cual la etapa clínica la determina el cirujano pediatra en la sala de operaciones y la confirma el patólogo. La clasificación, que se basa en el grado de extensión macroscópica y microscópica del tumor, es la misma para tumores con características histológicas favorables o desfavorables. Por lo tanto, los pacientes deberán ser caracterizados utilizando ambos criterios (18-19)
Etapa I (43% de los pacientes)
El Tumor de Wilms en etapa I esta definido como un tumor limitado al riñón y completamente extirpado. La superficie de la cápsula renal esta intacta y el tumor no se rompe ni antes de la escisión ni durante la misma. Los vasos del seno renal no están complicados y no hay tumor residual evidente mas allá de los márgenes de excisión.
Etapa II (23% de los pacientes)
El tumor de Wilms en etapa II se define como un tumor que se extiende mas allá del riñón pero que se extirpa completamente. No hay tumor residual evidente más allá de los márgenes de excisión. Se puede dar cualquiera de las siguientes condiciones:
1. Extensión regional del tumor, es decir, penetración a través de la superficie externa de la cápsula renal en el tejido blando perirenal o más de 1 a 2 mm de invasión tumoral en el seno renal.
2. Los vasos fuera del riñón están infiltrados o contienen trombo tumoral.
3. Se hizo una biopsia del tumor o hubo derramamiento local del tumor limitado al costado.
4. Durante el Estudio nacional del tumor de Wilms (NWTS-5), los tumores con indicios de invasión de los vasos en el seno renal (sin razón para ser clasificado como etapa lI)se clasificaron como etapa II en vez de la clasificación de etapa I a la que se les asignó durante NWTS-1 a 4. (20)
Etapa III (23% de los pacientes)
El Tumor de Wilms en etapa III se define como un tumor residual que está limitado al abdomen. Pueden darse una o varias de las siguientes condiciones:
1. Los ganglios linfáticos del hilio renal, de las cadenas periaórticas, o más allá resultan con tumores en la biopsia. La complicación de los ganglios linfáticos en el tórax u otros sitios extraabdominales seria un criterio para la etapa IV.
2. Ha habido contaminación peritoneal difusa por parte del tumor, como por derramamiento tumoral mas allá del flanco antes de la cirugía o durante la misma, o mediante crecimiento tumoral que ha penetrado la superficie peritoneal.
3. Se encuentran implantes en la superficie peritoneal.
4. El tumor se extiende más allá de los márgenes quirúrgicos microscópicos o macroscópicos.
5. El tumor no se puede resecar completamente debido a infiltración local en estructuras vitales.
Etapa IV (10% de los pacientes)
El Tumor de Wilms en etapa IV se define como la presencia de metástasis hematógena. Hay depósitos metastásicos más allá de la etapa III, por ejemplo, al pulmón, el hígado, los huesos, el cerebro o a una combinación de estos sitios.
Etapa V (5% de los pacientes)
El tumor de Wilms en etapa V se define como complicación renal bilateral en el momento del diagnostico inicial.
En los pacientes comprometidos de forma bilateral, se deberá intentar clasificar cada lado según los criterios anteriores en base al grado de la enfermedad antes de la biopsia. La supervivencia a 4 años era del 94% para aquellos pacientes cuya lesión más avanzada estaba en etapa I o II; y de 76% para aquellos cuya lesión mas avanzada estaba en etapa III. (21)
Anaplasia etapa I-IV (histología no favorable)
Los niños con tumores anaplásicos en etapa I tienen un pronóstico excelente. En su manejo puede emplearse el mismo régimen que se les da a los pacientes en etapa I de histología favorable. Los niños con anaplasia difusa en etapa II hasta la etapa IV, sin embargo, representan un grupo de mayor riesgo. Estos tumores son mucho más resistentes a la quimioterapia que tradicionalmente se administra a niños con tumor de Wilms (con histología favorable).(21).
En cuanto a los aspectos generales de las opciones de tratamiento, se ha establecido que la terapia consiste en cirugía seguida de quimioterapia y en algunos pacientes, radioterapia. (22-23).
El papel más importante del cirujano consiste en extraer el tumor en su totalidad sin romperlo y hacer una evaluación de la extensión de la enfermedad. El procedimiento a escoger consiste en obtener una muestra mediante nefrectomía radical de ganglios linfáticos a través de una incisión transabdominal con muestra de ganglios linfáticos En la actualidad no se recomienda la nefrectomía parcial. Aunque podría considerarse en tumores muy pequeños descubiertos mediante un tamizaje de ultrasonido. (24) E1 riñón contralateral debe palparse e inspeccionarse a través de una abertura en la fascia. Es mandatorio el obtener muestras de los ganglios linfáticos biliar, peri aórticos, iliacos y celiacos. Mas aun, debe tomarse muestra de cualquier ganglio sospechoso. Los márgenes de resecado, tumores residuales y cualquier recipiente de ganglio sospechoso debe ser marcado con ganchos sujetadores de titanio. Los resecados de cortes de hígado, o los resecados duodenales o colónicos parciales se aceptan para excisiones completas en bloque.
Los pacientes con tumores masivos unilaterales no resecables, tumores bilaterales o tumores trombóticos de vena cava por encima de las venas hepáticas, son candidatos a quimioterapia preoperatoria debido al riesgo de resección quirúrgica inicial.(25) La quimioterapia preoperatoria debe siempre seguir la biopsia (que puede ser llevada a cabo de forma percutánea) (26-31) La quimioterapia preoperatoria facilita la extracción del tumor y podría reducir la frecuencia con que se presentan las complicaciones quirúrgicas(25-31-32).
Los recién nacidos, así como todos los niños menores de 12 meses requieren una reducción del 50% de las dosis quimioterapéuticas administradas a niños mayores, (33) Esta reducción disminuye los efectos tóxicos que se manifestaron en niños de esta edad mientras sostuvieron unos resultados generales excelentes.(34) Los análisis sobre la función renal de los niños con Tumor de Wilms deben vigilarse cuidadosamente al principio de la terapia sobre la base de los efectos tóxicos renales(enfermedad veno oclusiva) de la que se ha reportado en estos pacientes.(35-36) Durante la radioterapia no debe administrase dactinomicina. Los niños con Tumor de Wilms tienen un riesgo creciente de desarrollar neoplasias malignos secundarios. Este riesgo depende de la intensidad de la terapia, incluso del uso de radiación y doxorrubicina y posibles factores genéticos. (37) En los niños que han sido sometidos a tratamiento con doxorubicina, se ha observado un aumento. El grado de riesgo esta influenciado por la dosis acumulativa de doxorubicina, irradiación al corazón y género (las mujeres corren un mayor riesgo.) (38) De forma que los esfuerzos están dirigidos a reducir la intensidad de la terapia siempre que sea posible. El pronóstico selección del tratamiento adicional para los pacientes con tumor de Wilms recurrente depende de muchos factores, incluyendo el sitio de recidiva, la histología del tumor, duración de la remisión inicial y de la quimioterapia.
Los pacientes con tumores de histología anaplásica desfavorable, recurrencia del tumor en el abdomen después de recibir tratamiento con radioterapia, recurrencia antes de pasar 6 meses de la nefrectomía o recurrencia después de una terapia inicial con 3 fármacos, tienen un pronóstico precario. La tasa de supervivencia en niños a 2 años después de una recurrencia local es de 43 % (40). La combinación de ifosfamida, etopósido y carboplatino ha demostrado actividad en este grupo de pacientes, pero se ha observado una toxicidad hematológica significativa. (41,42)
Referencias
1- Green DM, National Wilms,Tumor Study Group: Phase III Multimodality Therapy Based on Histology, Stage, Age, and Tumor Size in Chiidren With Wilms1 Tumor, Clear Cell Sarcoma of the Kidney, or Rhabdoid Tumors of the Kidney (Summary Last Modified 07/2002), NWTS-Q9401, clinical trial, active, 07/05/1995.
[ Links ]
2- Ritchey ML, Haase GM, Shochat S: Current management of Wilms' tumor. Seminars in Surgical Oncology 9(6): 502-509, 1993.
[ Links ]
3- Breslow N, Sharples K, Beckwith JB, et al.: Prognostic factors in nonmetastatic, favorable histology Wilms' tumor: results of the Third National Wilms1 Tumor Study. Cáncer 68(1): 2345-2353, 1991.
[ Links ]
4- Green DM, Breslow NE, Beckwith JB, et al.: Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Medical and Pediatric Oncology 21(3): 188-192, 1993.
[ Links ]
5- DeBaun MR, Siegel MJ, Choyke PL: Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome. Journal of Pediatrics 132 (3, pt 1): 401-404,1998.
[ Links ]
6- DeBaun MR, Tucker MA: Risk of cáncer during the first four years of lite in children from The Beckwith- Wiedemann Syndrome Registry. Journal of Pediatrics 132(3, pt 1): 398-400, 1998. [ Links ]
7- Porteus MH, Narkool P, Neuberg D, et al.: Characteristics and outcome of children with Beckwith-Wiedemann Syndrome and Wilms' tumor: a report from the National Wilms Tumor Study Group. Journal of Clinical Oncology 18(10): 2026-2031, 2000. [ Links ]
8- Hoyme HE, Seaver LH/ Jones KLÙ et al.: Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. American Journal of Medical Genetics 79(4): 274-278, 1998. [ Links ]
9- Clericuzio CL: Clinical phenotypes and Wilms tumor. Medical and Pediatric Oncology 21 (3): 182-187,1993.
[ Links ]
10- Diller L, Ghahremani M, Morgan J, et al.: Constitutional WT1 mutations in Wilms' tumor patients. Journal of Clinical Oncology 16(11): 3634-3640.
[ Links ]
11- Henry Anhalt, DO, E. Kirk Neely, MD, Raymond L Hintz, MD, Ambiguous genitalie. Pediatrics and Rev. VoI17.(6): 213-220. 1996.
[ Links ]
12- Bonaiti-Pellie C, Chompret A, Tournade MF, et al.: Genex-icü ciua epidemiology of Wilms' tumor: the French Wilms1 Tumor Study. Medical and Pediatric Oncology 20:(4): 284-291, 1992.
[ Links ]
13- Breslow NE, Beckwith JB: Epidemiological features of Wilms tumor: results of the National Wilms: Tumor Study. Journal of the National Cáncer Instituto 68(3): 429-436, 1982.
[ Links ]
14- Li FP, Williams WR, Gimbrere K, et al.: Heritable fraction of unilateral Wilms tumor. Pediatrics 81(1): 147- 149, 1988.
[ Links ]
15- Zuppan CW, Beckwith JB, Luckey DW: Anaplasia in unilateral Wilms' tumor: a report from the National Wilms' Tumor Study Pathology Center. Human Pathology 19(10): 1199-1209, 1988.
[ Links ]
16- Vujanic GM, Harms D, Sandstedt B, et al.: New definitions of focal and diffuse anaplasia in Wilms tumor: the International Society of Paediatric Oncology (SIOP) experience. Medical and Pediatric Oncology 32(5): 317-323, 1999.
[ Links ]
17- Faria P, Beckwith JB, Mishra K, et al.: Focal versus diffuse anaplasia in Wilms tumor-new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. American Journal of Surgical Pathology 20(8): 909-920, 1996.
[ Links ]
18- National Wilms: Tumor Study Committee: Wilms' tumor: status report, 1990. Journal of Clinical Oncology 9(5): 877-887, 1991.
[ Links ]
19- D'Angio GJ, Breslow N, Beckwith JB, et al.: Treatment of Wilms* tumor: results of the third National Wilms' Tumor Study. Cáncer 64(2): 349-360, 1989.
[ Links ]
20- Beckwith JB: National Wilms Tumor Study: an update for pathologists. Pediatric and Developmental Pathology 1 (1): 79-84, 1998.
[ Links ]
21- Ritchey ML, Pringle KC, Breslow NE, et al.: Management and outcome of inoperable Wilms tumor: a report of National Wilms Tumor Study-3. Aunáis of Surgery 220(5): 683-690, 1994.
[ Links ]
22- D'Angio GJ/ Breslow N, Beckwith JB, et al.: Treatment of Wilms' tumor: results of the third National Wilms' Tumor Study. Cáncer 64(2): 349-360, 1989.
[ Links ]
23- A. Jereb B, Burgers JM, Tournade MF, et al.: Radiotherapy in the SIOP (lnternational Society of Pediatric Oncology) nephroblastoma studies: a review. Medical and Pediatric Oncology 22(4): 221-227,1994.
[ Links ]
24- McNeil DE, Langer JC, Choyke P, et al.: Feasibility of partial nephrectomy for Wilm's tumor in children
with Beckwith-Wiedemann syndrome who have been screened with abdominal ultrasonography. Journal of Pediatric Surgery 37(1): 57-60, 2002.
[ Links ]
25- Ritchey ML: Primary nephrectomy for Wilms' tumor: approach of the National Wilms' Tumor Study Group. Urology 47(6): 787-791, 1996. [ Links ]
26- Tournade MF, Com-Nougue C, Voute PA, et al.: Results of the Sixth International Society of Pediatric Oncology Wilms' Tumor Trial and Study: a risk-adapted therapeutic approach in Wilms' tumor. Journal of Clinical Oncology 11 (6): 1014-1023, 1993.
[ Links ]
27- Oberholzer HF, Faikson G, De Jager LC: Successful management of inferior vena cava and right atrial nephroblastoma tumor thrombus with preoperative chemotherapy. Medical and Pediatric Oncology 20(1): 61- 63, 1992.
[ Links ]
28- Saarinen UM, Wikstrom S, Koskimies O, et al.: Percutaneous needle biopsy preceding preoperative chemotherapy in the management of massive renal tumors in children. Journal of Clinical Oncology 9(3): 406- 415, 1991. [ Links ]
29- Dykes EH, Marwaha RK, Dicks-Mireaux C, et al.: Risks and benefits of percutaneous biopsy and primary chemotherapy in advanced Wilms' tumour. Journal of Pediatric Surgery 26(5): 610-612,1991.
[ Links ]
30- Thompson WR, Newman K, Seibel N, et al.: A strategy for resection of Wilms' tumor with vena cava or atrial extensión. Journal of Pediatric Surgery 27(7): 912-915, 1992.
[ Links ]
31- Shamberger RC, Ritchey ML, Haase GM, et al.: Intravascular extensión of Wilms tumor. Annals of Surgery 234(1): 116-121. 2001.
[ Links ]
32- Shamberger RC, Guthrie KA, Ritchey ML, et al. Surgery-relatecTfactors and local recurrence of Wilms tumor in National Wilms Study 4. Armáis of Surgery 229(2): 292-297, 1999.
[ Links ]
33- Corn BW, Goldwein JW, Evans I, et al.: Outcomes in low-risk babies treated with halfdose chemotherapy according to the Third National Wilms' Tumor Study. Journai of Clinical Oncology 10(8): 1305-1309, 1992.
[ Links ]
34- Morgan E, Baum E, Breslow N, et al.: Chemotherapy-related toxicity in infants treated according to the Second National Wilms' Tumor Study. Journai of Clinical Oncology 6(1): 51-55,1988.
[ Links ]
35- Green DM/ Norkool P/ Breslow NE, et al.: Severe hepatic toxicity after treatment with vincristine and dactinomycin using singledose or divided-dose schedules: a report from the National Wilms' Tumor Study. Journai of Clinical Oncology 8(9): 1525-1530, 1990.
[ Links ]
36- Raine J, Bowman A, Wallendszusk K, et al.: Hepatopathy-thrombocytopenia syndrome - a complication of dactinomycin therapy for Wilms' tumor: a report from the United Kingdom Childrens Cáncer Study Group. Journai of Clinical Oncology 9(2): 268-273, 1991.
[ Links ]
37- Breslow NE, Takashima JR, Whitton JA, et al.: Second malignant neoplasms following treatment for Wilms' tumor: a report from the National Wilms' Tumor Study Group. Journai of Clinical Oncology 13(8): 1851-1859, 1995.
[ Links ]
38- Green DM, Grigoriev YA, Nan B, et al.: Congestive heart failure after treatment for Wilms' tumor: a report from the National Wilms: Tumor Study Group. Journai of Clinical Oncology 19(7): 1926-1934, 2001. [ Links ]
39- Grundy P, Breslow N, Green DM/ et al.: Prognostic factors for children with recurrent Wilms' tumor: results from the second and third National Wilms' Tumor Study. Journai of Clinical Oncology 7(5): 638-647.1989.
[ Links ]
40- Shamberger RC, Guthrie KA, Ritchey ML, et al.: Surgery-related facfcors and local recurrence of Wilms tumor in National Wilms Study 4. Aunáis of Surgery 229(2): 292-297, 1999. [ Links ]
41- Abu-Ghosh A, Goldman S, Krailo M/ et al.: Excellent responso rafe (91 %) to ifosfamide, carboplatin, and etoposide (ICE) in children with advanced and/or relapsed Wilms' tumor. Proceedings of the American Society of Clinical Oncoiogy A2157, 559a, 1999. [ Links ]
42- Kung FH, Bernstein ML/ Camitta BM, et al.: Ifosfamide/carboplatin/etoposide (ICE) in the treatment of advanced/ recurrent Wilms tumor. Proceedings of the American Society of Clinical Oncology A2156, 559a,1999. [ Links ]