










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Pediátrica Costarricense
Print version ISSN 1409-0090
Acta pediátr. costarric vol.18 n.1 San José Jan. 2004
Hepatoblastoma y Sarcoma embrionario: revisión y reporte de dos casos
David Romero Cubero 1, Luis A Corrales 2, Pedro Zúñiga 3 , Carlos Rodríguez 4
1 Médico General
2 Médico General, Servicio de Oncología, Hospital Nacional de Niños "Dr. Carlos Saénz Herrera"
3 Médico Residente de Pediatría
4 Pediatra Asistente, Servicio de Oncología, Hospital Nacional de Niños "Dr. Carlos Saénz Herrera"
Acta Pediátrica Costarricense 2004, volumen 17, número 3.
Caso 1
Se trata de un paciente masculino que a los 11 meses de edad, en una consulta de niño sano, el médico tratante le palpó una masa a nivel de hemiabdomen derecho por lo que fue referido al Hospital Nacional de Niños. Madre refiere que tenía 2 meses de irritabilidad, sin historia de pérdida de peso y sin ningún otro síntoma agregado.
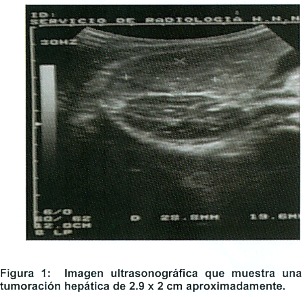
A su ingreso a emergencias el paciente se presentó afebril, hidratado con un abdomen blando, depresible y con una hepatomegalia de 4 cm bajo el reborde costal derecho. Se le realizaron una pruebas de función hepática, pruebas de coagulación y un control metabólico que se encontraban dentro de los limites normales. En su hemograma de ingreso presentaba una hemoglobina en 9,5 g/di, un leucograma normal, y 602.000 plaquetas. Se le realizó una deshidrogenasa láctica que se encontraba en 342 IU/L (N=91-180). Por su hepatomegalia se decide completar estudios con un ultrasonido abdominal (Figura 1) el cual reporta un hígado aumentado de tamaño con zona de 82 mm por 43 mm, de aspecto expansivo, heterogénea, mal delimitada que desplaza estructuras vasculares y el riñón derecho que involucra los segmentos 4, 5 y 6. Se le realiza un antígeno carcinoembrionario que se encuentra en 0,64 ng/ml (N= 0-20) y una alfa feto proteína con valores mayores a 300 (N= 0-20). La radiografía de tórax no muestra evidencia de patología. El paciente es ingresado para completar estudios entre los cuales se le realiza un TAC y posteriormente se decide realizar una biopsia por punción la cual se realiza a la semana de su internamiento y en la cual se reporta con el diagnóstico de hepatoblastoma. Posteriormente y durante el mismo internamiento, el paciente inicia quimioterapia con Cisplatinum, 5 Fluoracilo, y Vincristina.
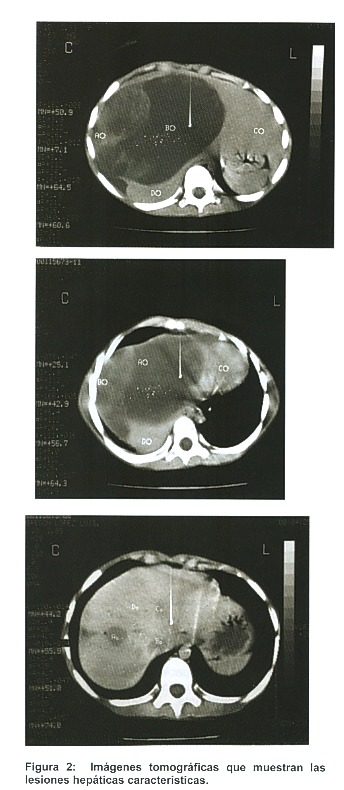
El paciente posteriormente recibe 2 ciclos más de quimioterapia (total de 3 ciclos) con los mismos antineoplásicos. En un nuevo TAC (Figura 2) se continúa observando la misma masa en hígado pero con una normalización de la alfa feto proteína (14 IU/L). El paciente es internado y a los 3 meses del diagnóstico se realiza una colecistectomía más hepatectomía derecha con bordes de resección reportados libres de neoplasia por patología.
A los 20 días de la cirugía el paciente recibe su ciclo de quimioterapia número 4, y ha evolucionado en forma satisfactoria con normalización de los valores de laboratorio pero mantiene su hemoglobina entre 9 g/dl y 10,5 g/dI. Actualmente se encuentra en control en Oncología y se deberá completar la quimioterapia.
Caso 2
Se trata de un paciente masculino que a los 6 años 9 meses inició con dolor abdominal difuso que cedía espontáneamente. No asociaba otra sintomatología. A los 2 meses de haberse iniciado el dolor, el paciente llega al Hospital Nacional de Niños alerta, conciente con un abdomen blando, depresible, sin signos de irritación peritoneal pero con una masa visible y palpable en hipocondrio derecho y epigastrio, de aproximadamente 8 cm de diámetro, dura, levemente dolorosa a la palpación. Por tal hallazgo se ingresa para completar estudios. En los exámenes de laboratorio el paciente presenta una hemoglobina en 9,4 g/dl, hematocrito 30%, con un tiempo de protrombina aumentado (porcentaje de actividad en 69%), albúmina en 2,5 (NI=3,2-5,5), AST y ALT normales, bilirrubinas normales, control metabólico normal, y antígeno carcinoembrionario normal.
Por tal motivo y al día siguiente de su ingreso se le realizó un ultrasonido que reportó una masa con múltiples áreas quísticas y áreas sólidas en el lóbulo hepático derecho de 15 cm por 14,7 cm por 14,4 cm. Con este hallazgo se le realiza una tomografía axial computarizada que reporta extensa lesión hepática heterogénea que compromete el lóbulo derecho del hígado de contornos definidos (Figuras 3 y 4). El paciente es llevado a sala de operaciones en donde se le realiza una laparotomía exploratoria con segmentectomía hepática derecha con resección tumoral completa y biopsia de la masa hepática la cual reporta un sarcoma hepático indiferenciado con extensa necrosis. Posteriormente el paciente inicia quimioterapia con Etopósido y Cisplatinum por un total de tres días en cada ciclo de tratamiento, recibiendo un total de cinco ciclos. Se le realiza un ultrasonido control después del tercer ciclo que muestra un nódulo de 4,5 cm por 3,5 cm por 3,5 cm, y un nuevo control de ultrasonido posterior al quinto ciclo de quimioterapia que no muestra una disminución de la masa. Por ende, se le realiza un nuevo TAC el cual muestra pequeñas áreas hipodensas a nivel del lóbulo hepático derecho (Figuras 5 y 6) por lo cual se decide ingresar para una nueva cirugía. Se le realiza, aproximadamente cinco meses después de su primer ingreso y posterior a su quinto ciclo de quimioterapia, una hepatectomía derecha con resección total macroscópica de la masa. La biopsia se reporta como negativa por neoplasia con bordes de resección libres. Posterior a la cirugía el paciente recibe su sexta y última dosis de quimioterapia. Actualmente está en control en Oncología del Hospital Nacional de Niños.
Discusión
Los tumores hepáticos primarios son poco comunes en la infancia, comprendiendo menos del 3% de los tumores de la edad pediátrica.(1) De estos tumores, aproximadamente dos tercios son benignos y un tercio malignos. (Tabla 1) (2) El hepatoblastoma es la malignidad hepática pediátrica primaria más común (comprende alrededor del 1 % de todos los cánceres pediátricos) seguida del carcinoma hepatocelular y más raramente los sarcomas hepáticos primarios. Con una incidencia de 0.7-1/1,000,000/año en países occidentales(3-5) Sin embargo hay que recordar que las lesiones hepáticas más comunes son aquellas producidas por metástasis de tumores como neuroblastoma, Wilms y linfomas.( 6 )
La edad de inicio del cáncer hepático en niños está relacionada con la histología del tumor. El hepatoblastoma ocurre generalmente antes de los 3 años de edad ( 7 ) La tasa de supervivencia general de los niños con este tumor es del 70% ( 8-10)
Existen diferentes subtipos histológicos de hepatoblastomas. Aproximadamente el 56% de los tumores son de tipo epitelial, el cual se puede subclasificar como fetal puro (31%), embrionario (19%), macrotrabecular (3%) e indiferenciado de células pequeñas (anaplásico 3%) y el restante 44% los comprenden tumores que contienen ambos componentes mixtos, tanto epitelial como mesenquimatoso tipo ostioide o cartílago.(11)
Un análisis de pacientes con hepatoblastoma resecable ha sugerido que los tumores que se caracterizan por tener una histología puramente "fetal" tienen un pronóstico mejor que aquellos que tienen un agregado de componentes embrionarios más primitivos y de división rápida u otros tejidos no diferenciados. Suelen presentarse como masas bien delimitadas de 10 a 12 cm en el momento del diagnóstico. (12-14)
La característica de presentación más común de las neoplasias hepáticas pediátricas es la presencia de una masa abdominal asintomática.(1, 2). Dentro de los síntomas y signos que suelen presentar más comúnmente los niños con hepatoblastomas además se pueden encontrar: pérdida de peso, hiporexia , dolor abdominal, vómitos, ictericia, fiebre, prurito, palidez producto de la anemia, dolor lumbar por compresión del tumor, entre otros. Sin embargo estos pueden variar según tamaño del tumor y la presencia y ubicación de las metástasis. (4, 5, 13)
Ocasionalmente los hepatoblastomas producen gonadotropina coriónica beta_humana que produce como resultado la precocidad isosexual. La osteopenia severa es común. El hepatoblastoma es parte de la constelación de hallazgos asociados con el síndrome de Beckwith_Wiedemann.(16) Así que dicha anormalidad genética podría estar directamente involucrada en algunos casos en la patogénesis del hepatoblastoma. (17,18) Alrededor de 2% de los niños que padecen de hepatoblastoma tienen hemihipertrofia. (19) Menos del 1 % de los niños con hemihipertrofía presentan un aumento en el riesgo de desarrollar hepatoblastoma durante los primeros años de vida.(20) Existe además una asociación clara entre el hepatoblastoma y la poliposis adenomatosa familiar (FAP), por lo que los niños de familias portadoras del gen FAP corren un riesgo mayor de padecer un hepatoblastoma, aunque éste ocurre en menos del 1 % de los miembros de la familia que tiene FAP.(21-23)También se ha informado de la existencia de una relación entre los niños de peso bajo al nacer y los hepatoblastomas.(24-26) Hasta un 10% de los pacientes con hepatoblastoma tienen historia de prematuridad con hospitalización prolongada.(27) A diferencia del carcinoma hepatocelular el hepatoblastoma no guarda relación con la cirrosis.( 5)
La mayoría de los pacientes con hepatoblastoma (más del 90%) tienen un marcador tumoral sérico, alfa_fetoproteína elevado, que refleja en forma paralela la actividad de la enfermedad. La falta de disminución significativa en los niveles de alfa_fetoproteína con el tratamiento puede predecir una mala respuesta a la terapia.(28) Aquellos pacientes con niveles de AFP normales o bajos o por el contrario extremadamente elevados tienen un pobre pronóstico en contraste con aquellos que presentan valores intermedios. En un estudio, pacientes con niveles bajos presentaron la variante de células pequeñas, que con frecuencia no produce AFP, crecen rápidamente y no suelen responder a quimioterapia. Niveles extremadamente altos de AFP se asociaron con tumores extensos y/o metastásicos y por lo tanto un desarrollo desfavorable. (29)
La resecabilidad quirúrgica es el factor de sobrevida más importante para pacientes con hepatoblastoma. Sin embargo la resección hepática es posible solo en una minoría de pacientes. Típicamente estos tumores se consideran irresecables cuando compromete tanto el lóbulo derecho como el izquierdo del hígado, cuando involucra las venas hepáticas o la vena cava inferior; si la enfermedad es multifocal difusa o cuando el tamaño del tumor es extremadamente grande y puede resultar en sangrado excesivo. (30)
La tasa de resecabilidad ha mejorado dramáticamente con la quimioterapia. El tratamiento del hepatoblastoma metastásico avanzado irresecable ha mejorado pero sigue siendo insatisfactorio con tasas de sobrevida para etapas 3 y 4 del 65% y 15% respectivamente.(31,32) La quimioembolización y/o el transplante hepático están siendo evaluados para tratar la enfermedad irresecable localizada en el hígado.( 33,34 )
La mayoría de los pacientes sobrevive a la extracción de un hepatoblastoma que ha sido extraído por completo, pero sólo una minoría de los pacientes tiene lesiones receptivas a una resección completa en el momento del diagnóstico. La imposibilidad de extraer completamente el tumor primario o la presencia de enfermedad metastática se asocia con un resultado precario. El hepatoblastoma suele ser unifocal, mientras que el carcinoma hepatocelular con frecuencia es ampliamente invasor o multicéntrico. Por lo tanto, la resección es posible con más frecuencia en el hepatoblastoma que en el carcinoma hepatocelular, en el cual menos del 30% es resecable.( 5 )
Clasificación posquirúrgica de las etapas
Para agrupar a los niños con cáncer hepático, en los Estados Unidos se ha empleado un sistema de clasificación basado en la extensión postquirúrgica del tumor y en la posibilidad de resección quirúrgica. Este sistema de clasificación se emplea para determinar el tratamiento.( 36,37 ) Los niños diagnosticados con hepatoblastomas en etapa I y II tienen una tasa de curación mayor del 90% en comparación con el 60% que representa la etapa III y el 20% que representa la etapa IV.
Etapa I No hay metástasis, completamente resecado.
Etapa II No hay metástasis, tumor resecado de forma macroscópica con enfermedad residual microscópica (es decir, márgenes positivos); ruptura del tumor o tumor esparcido durante la cirugía.
Etapa III No hay metástasis distantes, tumor irresecable o resecable con tumor residual macroscópico o ganglios linfáticos positivos.
Etapa IV Hay metástasis distantes sin importar el grado de complicación hepática.
Clasificación prequirúrgica de las etapas mediante el uso de técnicas de imagenología
Categoriza el tumor primario con base en la extensión del compromiso hepático por cuadrantes al momento del diagnóstico. La supervivencia general de 5 años que se logró en este estudio internacional en que los niños se trataron con quimioterapia preoperatoria con doxorubicina y cisplatino fue de 100%, 91%, 68%, y 57% en las etapas de 1 a 4 respectivamente, incluyendo a pacientes con metástasis.( 38, 39 )
PRETEXT etapa 1 El tumor se ha extendido a solo un cuadrante, y los tres cuadrantes restantes del hígado, están exentos de tumor.
PRETEXT etapa 2 El tumor se extiende a dos cuadrantes adjuntos y los dos cuadrantes restantes están exentos de tumor.
PRETEXT etapa 3 El tumor se extiende a tres cuadrantes adjuntos o a dos cuadrantes no adjuntos, y un cuadrante solo, o dos cuadrantes no adjuntos, están exentos de tumor.
PRETEXT etapa 4 El tumor se extiende a los cuatro cuadrantes; no hay cuadrantes exentos de tumor.
El tratamiento del hepatoblatoma clasificado postquirúrgicamente en etapas I y II y prequirúrgica PRETEXT 2 y 3 se basa en quimioterapia de combinación antes del intento de la extracción quirúrgica del tumor (36). El PRETEXT 1 se debe resecar sin quimioterapia previa. En estos casos se logra una sobrevida mayor al 90%.
Los tumores clasificados como etapa III postquirúrgica y prequirúrgica PRETEXT 4 inicialmente no resecable, puede volverse resecable con quimioterapia preoperatoria con cisplatino logrando sobrevida libre de enfermedad de un 60 a 65% (37)
En la etapa 4 postquirúrgica puede lograrse curación del 25-30% con quimioterapia preoperatoria, resección del tumor primario y cualquier residuo de metástasis pulmonar. ( 36,37 )
Los pacientes en etapas 3 y 4 de la clasificación postquirúrgica, cuyos tumores permanecen irresecables deben ser considerados para quimioterapia alternativa como dosis altas de cisplatino con etopósido (37), radioterapia (38), infusión hepática directa de agentes quimioterapéuticos (39) o transplante ortotópico hepático.(40)
Sarcoma embrionario
Este tumor fue identificado como entidad única en 1978 por Stocker y SAC.(41) Se presenta en niños con edades entre 5 y 10 años (41,42) y no muestra predilección por género.(41) Representa el 6% de todos los tumores hepáticos en niños y el 13% de las neoplasias hepáticas malignas en este grupo etario. La sobrevida a largo plazo libre de enfermedad fue obtenida solo en pacientes en que se pudo resecar completamente el tumor.(42)
Suele ser un tumor bien delimitado del hígado normal, sin embargo a menudo da infiltración de elementos malignos más allá de la aparente pseudocápsula. (42)
Histopatológicamente muestra elementos mesenquimatosos malignos sin ninguna evidencia de diferenciación específica. Dicha variabilidad histológica dificulta su diagnóstico y da origen a gran variedad de nombres con que se conoce como: mesenquimoma maligno, rabdomiosarcoma del hígado, fibromiosarcoma, sarcoma indiferenciado o sarcoma embrionario. Se cree que puede haber relación entre el hamartoma y el sarcoma así como con el hepatoblastoma.(42)
Clínicamente suele presentarse como una masa abdominal que puede acompañarse de dolor y en algunos casos por síntomas sistémicos como fiebre, pérdida de peso o vómito.(41,43) A la evaluación angiográfica la apariencia es avascular o hipovascular. La ultrasonografía y la tomografía axial demuestran el aspecto sólido y quístico observado macroscópicamente.(43)
La inmunohistoquímica positiva para vimentina y alfa 1 antitripsina u otros antígenos mesenquimatosos está comúnmente presente.(43)
La quimioterapia preoperatoria puede producir una disminución suficiente del tumor de manera tal que facilite la resección quirúrgica. Se había considerado una neoplasia de pronóstico desfavorable, sin embargo gracias a tratamiento multimodal moderno que incluye resección quirúgica completa, radioterapia, quimioterapia y tratamiento de soporte la sobrevida de estos paciente ha mejorado considerablemente.(41-43)
Dado que no existe tratamiento estándar hasta la fecha, el grupo cooperativo italiano-alemán independientemente decidieron tratarlos de acuerdo con las guías diseñadas para niños con rabdomiosarcoma.(41)
A pesar de ser un tumor quimiosensible no hay evidencia que la quimioterapia sola o en combinación con radioterapia puedan curarlo.(42)La experiencia en cuanto a transplante hepático en este tipo de patología es limitada, sin embargo debe ser considerado luego de quimioterapia cuando la cirugía completa no es factible luego de una extensa búsqueda de metástasis regionales o distantes.(41) Pese a la resecabilidad completa en algunos casos las recurrencias locales y metástasis distantes han sido el mayor impedimento para obtener una sobrevida a largo plazo libre de enfermedad. (42)
Agradecimientos
Se agradece al Dr. José Carlos Barrantes, Cirujano Oncólogo del Hospital Nacional de Niños por la revisión crítica de este manuscrito.
Referencias
1. Gurarangan S, O'Meara A, MacMahon C, Guiney EF, O'Donnell B, Fitzgerald RJ, Breatnach F: Primary hepatic tumors in children: a 26 year review. J Surg Oncol 1992, 50:30-36. [ Links ]
2. Weinberg AG, Finegold MJ: Primary hepatic tumors of childhood. Hum Pathol 1983, 14:512. [ Links ]
3. Herzog CE, Andrassy RJ, Eftekhari F: Childhood cancers: hepatoblastoma. Oncologist 2000; 5: 445-53. [ Links ]
4. Newman KO: Hepatic tumors in children. Semin Pediatr Surg 1997; 6: 38-41. [ Links ]
5. Reynolds, M. : Pediatric liver tumors. Semin Surg Oncol. 1999;16:159-72. [ Links ]
6. Stocker JT: Hepatic tumors in children. Clin Liver Dis. 2001 ; 5: 259-81. [ Links ]
7. Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub. No. 994649. [ Links ]
8. Ortega JA, Krailo MO, Haas JE, et al.: Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Childrens Cancer Study Group. J Clin Oncol 9: 2167-76,1991. [ Links ]
9. Douglass EC, Reynolds M, Finegold M, et al.: Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol 11: 96-9, 1993. [ Links ]
10. Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/ vincristine/fluorouracil and cisplatin/ continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin OncoI2000;18: 2665-75,. [ Links ]
11. Williams RA, Ferrell LO: Pediatric liver tumors. Pathology (Phila) 1993; 2: 23-42. [ Links ]
12. Ishak KG, Glunz PR: Hepatoblastoma and hepatocarcinoma in infancy and childhood. Report of 47 cases. Cancer 1967;20: 396-422,. [ Links ]
13. Weinberg AG, Finegold MJ: Primary hepatic tumors of childhood. Hum Pathol 1983;14 : 512-37. [ Links ]
14. Haas JE, Muczynski KA, Krailo M, et al.: Histopathology and prognosis in childhood hepatoblastoma and hepatocarcinoma. Cancer 1989; 64: 1082-95. [ Links ]
15. Bhattacharya S, Lobo FO, Pai PK: Hepatic neoplasms in childhood - a clinicopathologic study. Pediatr Surg Int 1998; 14: 51-54. [ Links ]
16. Sotelo-Avila C, Gonzalez-Crussi F, Fowler JW: Complete and incomplete forms of Beckwith-Wiedemann syndrome: their oncogenic potential. J Pediatr 1980;96: 47-50. [ Links ]
17. Albrecht S, von Schweinitz O, Waha A, et al.: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 1994;54: 5041-4. [ Links ]
18. Mannens M, Hoovers JM, Redeker E, et al.: Parental imprinting of human chromosome region 11 p15.3-pter involved in the Beckwith-Wiedemann syndrome and various human neoplasia. Eur J Hum Genet 1994; 2 3-23. [ Links ]
19. Fraumeni JF Jr, Miller RW, HiII JA: Primary carcinoma of the liver in childhood: an epidemiologic study. J Natl Cancer Inst 40: 108799, 1968. [ Links ]
20. Hoyme HE, Seaver LH, Jones KL, et al.:lsolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet 79: 2748, 1998. [ Links ]
21. Iwama T, Mishima Y: Mortality in young first-degree relatives of patients with familial adenomatous polyposis. Cancer 73: 2065-8, 1994. [ Links ]
22. Li FP, Thurber WA, Seddon J, et al.: Hepatoblastoma in families with polyposis coli. JAMA 257: 2475-7,1987. [ Links ]
23. Garber JE, Li FP, Kingston JE, et al.: Hepatoblastoma and familial adenomatous polyposis. J Natl Cancer Inst 80: 1626-8, 1988. [ Links ]
24. Koch A, Oenkhaus O, Albrecht S, et al.: Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the betacatenin gene. Cancer Res 59: 269-73, 1999. [ Links ]
25. Ikeda H, Hachitanda Y, Tanimura M, et al.: Development of unfavorable hepatoblastoma in children of very low birth weight: results of a surgical and pathologic review. Cancer 82: 178996, 1998. [ Links ]
26. Maruyama K, Ikeda H, Koizumi T, et al.: Prenatal and postnatal histories of very low birthweight infants who developed hepatoblastoma. Pediatr Int 41: 82-9, 1999. [ Links ]
27. Tanimura M, Matsui 1, Abe J: Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res 1998 15; 58: 3032-5.
28. Van Tornout JM, Buckly JO, Quinn JJ, Feusner JH, Krailo MO, King OR, Hammond GO, Ortega JA: Timing and magnitude decline in alphafetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: a report from the Children's Cancer Group. J Clin Oncal 1997, 15:1190-1197. [ Links ]
29. Van Tornout JM, Buckley JO, Quinn JJ: Timing and magnitude of decline in alpha-fetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: a report from the Children's Cancer Group. J Clin Oncol1997 Mar; 15(3): 1190-1197. [ Links ]
30. Achilleos OA, Buist LJ, Kelly DA: Unresectable hepatic tumors in childhood and the role of liver transplantation. J Pediatr Surg 1996 Nov; 31: 1563-1567. [ Links ]
31. Van Schweinitz O, Hecker H, Harms O, Bode U, Weinel P, Burger O, Erttmann R, Mildenberger H: Complete resection before development of drug resistance is essential for survival from the advanced hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB-89. J Pediatr Surg 1995, 30:845-852. [ Links ]
32. Plaschkes J, Perilongo G, Shafford EA, Brock P, Brown J, Oicks-Mireaux C, Habrand JL, Keeling J, Philips A, Pritchard J, Vos A: SIOP trial report: overall preliminary results of SIOPEL-I for the treatment of hepatoblastoma (HB) with preoperative chemotherapy. Continuous infusion cisplatin and doxrubicin (PLAO). Med Pediatr Oncal 1994, 23: 170. [ Links ]
33. Koneru B, Flye MW, Busuttil RW, Shaw BW, Lorber MI, Emond JC, Kalayogulu M, Freese OK, Starzl TE: Liver transplantation for hepatoblastoma: the American experience. Ann Surg 1991, 213:118-121. [ Links ]
34. Oue T, Fukuzama M, Kusafuka T, Kohmoto Y, Okada A, Imura K: Transcatheter arterial chemoembolization in the treatment of hepatoblastoma. J Pediatr Surg 1998, 33:17711775. [ Links ]
35. Exelby PR, Filler RM, Grosfeld JL: Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular
carcinoma: American Academy of Pediatrics Surgical Section Survey--1974. J Pediatr Surg 10: 329-37, 1975.
36. Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin / continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin OncoI2000;18: 2665-75. [ Links ]
37. Douglass E, Ortega J, Feusner J, et al.: Hepatocellular carcinoma (HCA) in children and adolescents: results from the Pediatric Intergroup Hepatoma Study (CCG 8881/POG 8945). [Abstract] Proceedings Am Soc Clin Oncology 1994;13:1439- 420.
38. Pritchard J, Brown J, Shafford E, et al.: Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: a successful approach--results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol 2000;18:3819-28. [ Links ]
39. Brown J, Perilongo G, Shafford E, et al.: Pretreatment prognostic factors for children with hepatoblastoma-- results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer 2000; 36: 1418-25. [ Links ]
40. Reyes JO, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 2000; 136: 795-804,. [ Links ]
41. Bisogno G, Pilz T, et al.:Undifferentiated Sarcoma of the Liver in Childhood a curable disease. Cancer 2002; 94: 252-257. [ Links ]
42. Urban Ch, Mache C, et al.: Undifferentiated (Embryonal) Sarcoma of the Liver in Childhood. Cancer 1993; 72: 2511-2516,. [ Links ]
43. Webber E, Morrison K, et al.: Undifferentiated Sarcoma of the Liver. J Pediatr Surg 1999;34:1641-1644. [ Links ]
