










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Pediátrica Costarricense
Print version ISSN 1409-0090
Acta pediátr. costarric vol.17 n.2 San José Jan. 2003
Carlos Rodríguez Rodríguez1, David Romero Cubero2, Pedro Zúñiga Arias3, Luis Alonso Picado Rodríguez4
1 Pediatra,
2 Médico General,
3 Médico Residente de Pediatría,
4 Médico Residente de Pediatría
Dirección: Servicio de Oncología, Hospital Nacional de Niños. Teléfono: 839 0951 Correo electrónico: carlosro2@hotmail.com
Acta Pediátrica Costarricense 2003, volumen 17, número 2
Resumen
Los autores presentan el caso de un paciente de 2 años con historia de ser portador de retinoblastoma bilateral diagnosticado en octubre del 2001. Recibió N° 6 ciclos de quimioterapia y no hubo respuesta adecuada. Fue tratado con cirugía láser en Colombia y luego recibió Nº2 ciclos más de quimioterapia. Actualmente consulta por tumoración en ojo izquierdo. Se le realiza TAC de órbita que muestra masa de tejidos blandos que proyecta el globo ocular hacia adelante y persistencia de calcificaciones previas.
Palabras Claves: retinoblastoma, germinolineal, esporádico, enucleación.
Caso
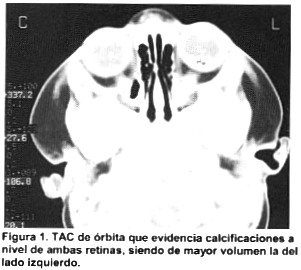
Se trata de un paciente de 2 años, vecino de Liberia que fue diagnosticado portador de retinoblastoma bilateral. Fue tratado con Nº 6 ciclos de quimioterapia sin una respuesta adecuada. Valorado por oftalmología quienes plantean enucleación de ojo izquierdo y quimioterapia para el ojo derecho. Se le realiza TAC de órbita donde se aprecian calcificaciones a nivel de ambas retinas, siendo de mayor volumen la del lado izquierdo. (Figura 1).
Debido a la negativa de los padres para tratamiento quirúrgico, recibió cirugía láser en Colombia, así como Nº2 nuevos ciclos de quimioterapia. Actualmente consulta por tumoración importante a nivel de ojo izquierdo.
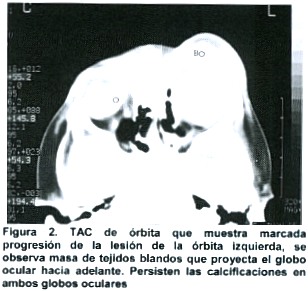
Se le realiza TAC de órbita que muestra marcada progresión de la lesión de la órbita izquierda, observándose masa de tejidos blandos que proyecta el globo ocular hacia adelante. Persisten en ambos globos oculares las calcificaciones. (Figura 2).
Se decide en conjunto por los servicios de Oncología y Oftalmología realizar un nuevo ciclo de quimioterapia y posterior enucleación del ojo izquierdo.
Discusión
El retinoblastoma es un tumor relativamente común en la niñez que aparece en la retina y representa alrededor del 3% de los cánceres que surgen entre los niños menores de 15 años de edad ( 1 ). La retina es la capa más profunda del ojo y se encuentra en su parte posterior. Es la parte del ojo que recibe la luz y las imágenes necesarias para la visión.
Se estima que la incidencia anual es de alrededor de 4 casos por cada millón de niños. Aunque puede ocurrir a cualquier edad, se presenta con mayor frecuencia en preescolares con un 95% de los casos diagnosticados antes de la edad de 5 años. La presentación y el curso clínico de los pacientes mayores de 5 años de edad pueden diferir de los pacientes más jóvenes( 2 ). El tumor puede ser unilateral (75%) o bilateral (25%). El retinoblastoma se limita generalmente al ojo y como resultado, 90% de los niños con retinoblastoma tienen una tasa de supervivencia de 5 años que sobrepasa el 90%.
El retinoblastoma es un tumor que se presenta como línea germinal (40%) o esporádico (60%). La enfermedad germinolineal comprende aquellos pacientes con historial familiar con individuos positivos a esta enfermedad (hereditaria) y aquellos pacientes que han heredado una mutación germinolineal de uno de los padres no afectados.
El retinoblastoma se produce a causa de la mutación de un gen supresor de tumores denominado RB1, que se encuentra en el cromosoma 13. Se necesitan 2 mutaciones ( cambios en un gen) para destruir este gen y causar el crecimiento descontrolado de células. Cuando el retinoblastoma es hereditario (40% de los casos), la primera mutación se hereda de uno de los padres, mientras que la segunda se produce durante el desarrollo de la retina. Por otro lado, cuando el retinoblastoma es esporádico (60% de los casos) ambas mutaciones se producen durante el desarrollo de la retina. Esporádico significa" que ocurre por azar". También se han descubierto alteraciones en el gen RB1 en otros tumores, entre los que se encuentran el osteosarcoma y el cáncer de mama.
En general, los tumores en la mayoría de los niños con retinoblastoma hereditario perjudica a los dos ojos ( en efecto, todos los casos en que ambos ojos se ven afectados se deberían considerar hereditarios). El gen RB1 es un gen autosómico dominante. Esto significa que ambos sexos se ven afectados en igual medida y que en cada embarazo existe un 50% de posibilidad de que uno de los padres transmita el gen a su hijo. Cuando el hijo hereda el gen, existe entre un 75% y un 90% de posibilidades de que se produzca una segunda mutación, la cual da origen al retinoblastoma. Por lo tanto, es posible que un niño que hereda la mutación no experimente la segunda mutación, y en consecuencia, nunca desarrolle el retinoblastoma (3, 4).
Se recomienda que los niños sean examinados cada 2 a 4 meses por al menos 28 meses( 5 ). Después del tratamiento, los pacientes requieren de un vigilancia cuidadosa hasta cumplir 7 años de edad( 6 ). El retinoblastoma trilateral es un síndrome bien reconocido que consiste en retinoblastoma germinolineal unilateral o bilateral asociado a un tumor neuroblástico intracraneal. Se ha observado que entre el 5 y el 15% de los niños ya sea con retinoblastoma familiar, multifocal o bilateral podrían desarrollar de igual manera, un tumor neuroblástico intracraneal( 7 ). Los niños con retinoblastoma germinolineal tienen particularmente una alta incidencia de retinoblastoma trilateral, el cual es casi siempre mortal. También se ha descubierto que los pacientes que son asintomáticos al momento de ser diagnosticados con tumor intracraneal, gozan de una mejor supervivencia en sentido general, que aquellos pacientes sintomáticos( 7 ). Los exámenes de detección mediante neuroimagenología podrían mejorar la tasa de curación si los casos de retinoblastoma trilateral se detectan durante el primer año después del diagnóstico. Se ha recomendado que a los niños con retinoblastoma germinolineal se les practique un examen de detección en el que se use resonancia magnética cada 3 meses durante el primer año después del diagnóstico, y al menos 2 veces por año durante los 3 años siguientes( 8 ).
Cualquier individuo con antecedentes familiares positivos de retinoblastoma debe solicitar asesoría genética para identificar los riesgos específicos de transmisión del gen o enfermedad en los niños. La consejería de tipo genética debe ser una parte integral de la terapia de paciente con retinoblastoma, ya sea unilateral o bilateral. Sin embargo, este tipo de consejería no siempre es específica. Aquellas familias con retinoblastoma podrían tener un precursor con mutagenicidad embriónica que causa mosaisismo genético de gametos( 9 ). Una porción significante (10 a 18%) de niños con retinoblastoma tiene mosaisismo genético somático( 10 ) logrando hacer la historia genética algo más complejo y contribuir a la dificultad de la consejería genética.
Los síntomas más frecuentes del retinoblastoma pueden variar en cada paciente, y podrían incluir leucocoria, estrabismo, dolor o enrojecimiento ocular uni o bilateral y cambios en la agudeza visual del niño. A menudo, el diagnóstico precoz de la enfermedad puede evitar que los síntomas del retinoblastoma se manifiesten. Estos síntomas pueden parecerse a los de otras condiciones o problemas médicos. Además del examen físico o la historia clínica, los procedimientos para diagnosticar el retinoblastoma pueden incluir examen ocular completo, fondo de ojo, TAC, resonancia magnética nuclear, análisis sanguíneos, análisis de líquido cefalorraquídeo y médula ósea, y exámenes genéticos o de ADN. El tipo de tratamiento requerido depende tanto del grado de la enfermedad dentro del ojo, como de si la enfermedad se ha diseminado más allá del ojo, ya sea el cerebro o al resto del cuerpo ( 11 ).
La mayoría de los pacientes con retinoblastoma presentan enfermedad extensa dentro del ojo al momento del diagnóstico, con tumores masivos que afectan más de la mitad de la retina, tumores múltiples que afectan difusamente la retina, o impregnación obvia del humor vítreo. La meta de la terapia tiene dos propósitos: Curar la enfermedad y preservar la visión. El tumor está compuesto principalmente de células anaplásicas indiferenciadas que surgen de las capas nucleares de la retina. La histología muestra una semejanza al neuroblastoma y al meduloblastoma que incluye agregación alrededor de los vasos sanguíneos, necrosis, calcificación y las rosetas de Hexner-Wintersteiner. Los retinoblastomas están caracterizados por la proliferación de células marcadas con evidencia de un conteo alto de mitosis e índices extremadamente altos de los marcadores MIB-1( 12 ).
A pesar de que existen varios sistemas de clasificación actualmente disponibles para retinoblastoma, para los fines de tratamiento, el retinoblastoma se categoriza como enfermedad intraocular y extraocular. El retinoblastoma intraocular se localiza en el ojo y puede limitarse a la retina o puede extenderse afectando el globo; sin embargo, no se extiende más allá del ojo en los tejidos alrededor del ojo o a otras partes del cuerpo. La supervivencia libre de enfermedad a 5 años plazo es mayor al 90%.
El retinoblastoma extraocular se ve extendido más allá del ojo, puede limitarse a tejidos alrededor del ojo o puede haberse diseminado tipicamente al sistema nervioso central o a otras partes del cuerpo. La supervivencia libre de enfermedad a 5 años plazo es menor del 1 0%.
Clasificación de Reese-Ellsworth para tumores intraoculares
Reese y Ellsworth desarrollaron una clasificación generalmente adoptada de retinoblastoma intraocular y que ha demostrado tener importancia pronóstica en el mantenimiento de la vista y el control de la enfermedad local. El sistema se considera de importancia en las decisiones referentes al uso de modalidades locales de tratamiento.
Grupo II: favorable para la conservación de la vista
Grupo III: posible conservación de la vista
Grupo IV: desfavorable para la conservación de la vista
Grupo V: muy desfavorable para la conservación de la visión
Aproximadamente 90% de los pacientes presentan enfermedad con uno o ambos ojos categorizados en el grupo V.
Sistema Clínico de Clasificación del St. Jude Children's Research Hospital
Etapa II: tumor limitado al globo
Etapa III: extensión regional extraocular del tumor
Etapa IV: metástasis distantes
Aproximadamente el 80% de los pacientes presentan enfermedad en uno o ambos ojos clasificados en etapas I-II. El tratamiento específico para el retinoblastoma será determinado según la edad del paciente, su estado general de salud, historia médica, estadio de la enfermedad y expectativas para la evolución de la enfermedad. El principal objetivo del tratamiento consiste en extirpar el tumor y evitar la metástasis. Se requiere la planificación del tratamiento por un equipo multidisciplinario de especialistas en cáncer que tienen experiencia para tratar tumores oculares infantiles para determinar e implementar un tratamiento óptimo. Debido a la complejidad de la terapia, la pericia en radioterapia pediátrica y en oftalmología deberían estar a disposición. EI tratamiento para el retinoblastoma intraocular debería planificarse después que se conozca el grado tumoral dentro y fuera del ojo. Las opciones de tratamiento consideran tanto la curación como la preservación de la vista.( 15 )
Las opciones de tratamiento para el ojo afectado son: Enucleación, si el tumor es masivo o si hay
poca expectativa de mantener visión útil, radiación con haz externo, crioterapia, ocasionalmente, se emplea sola la coagulación por luz (fotocoagulación) en tumores pequeños. ( 16 ) Braquiterapia con placas radiactivas para las presentaciones focales unilaterales o para enfermedad recurrente después de irradiación previa con haz externo. ( 17 ) Aún está bajo investigación, el uso de quimioterapia sistémica para reducir volumen del tumor (quimioreducción) y para eludir los efectos a largo plazo ocasionados por la radioterapia en pacientes con tumores intraoculares que no son tratables mediante crioterapia, o fotocoagulación sola. ( 15,18 )
Los pacientes con un solo ojo afectado, generalmente se encuentra que tienen afección masiva de ese ojo. Puesto que la mayoría de las enfermedades unilaterales son generalmente masivas a menudo no hay ninguna expectativa de preservar la visión, usualmente se inicia cirugía ( enucleación) y no se administra radioterapia a la base del tumor. Sin embargo en los casos donde existe la posibilidad de preservar la visión debido a que los tumores son más pequeños, deberá considerarse tratamiento con otras modalidades (radioterapia, fotocoagulación, crioterapia, termoterapia, quimiorreducción y braquiterapia) en vez de cirugía. Ya que una proporción de niños que presentan retinoblastoma unilateral con el tiempo desarrollan la misma enfermedad en el ojo opuesto, es muy importante que los niños con retinoblastoma unilateral sean examinados periódicamente del ojo no afectado. La enfermedad bilateral asincrónica ocurre más frecuentemente en familias en que los padres están afectados.
El manejo de la enfermedad bilateral depende del grado de la enfermedad en cada ojo. Generalmente la enfermedad se encuentra más avanzada en un ojo, con menos afección del otro ojo. En el pasado el tratamiento estándar era la extirpación del globo ocular con mayor daño, sin embargo, si existe la posibilidad de preservar la visión en ambos ojos se indica efectuar irradiación bilateral prestando una particular atención a la respuesta. Un número reducido de pacientes con retinoblastoma presentan enfermedad extraocular. La cual puede localizarse en los tejidos blandos que rodean el ojo o en el nervio óptico más allá del margen de resección. Sin embargo puede ocurrir extensión adicional al cerebro y meninges con sembrado posterior en el líquido espinal, así como enfermedad metastásica distante que afecta los pulmones, huesos y médula ósea.
No existe una terapia efectiva claramente probada para el tratamiento del retinoblastoma extraocular, aunque se han usado la irradiación orbital y la quimioterapia. En general, se ha usado terapia paliativa con radiación (incluyendo irradiación craneoespinal cuando hay afección meníngea) o quimioterapia con metrotexato, citarabina e hidrocortisona junto con asistencia médica de apoyo al paciente. ( 19 )
El pronóstico para un paciente con retinoblastoma recidivante o progresivo depende del sitio y del grado de la recidiva o progresión. Se ha informado de respuestas tan elevadas como un 85% después de tratamiento con etopósido y carboplatino . Si la recidiva o la progresión del retinoblastoma se limita al ojo y es pequeña, el pronóstico para la vista y la supervivencia serán excelentes solo con terapia local, mientras que si se limitan al ojo pero son extensas, el pronóstico para la vista es pobre, no obstante la supervivencia continúa siendo excelente; si es extraocular el pronóstico empeora notablemente.
El seguimiento continuo es esencial para un niño al que se le diagnostica un retinoblastoma. Los cánceres secundarios tienen una gran incidencia en los pacientes que sobreviven al retinoblastoma. Estos cánceres no constituyen una recaída o recurrencia del retinoblastoma, sino que son tumores primarios de otros órganos. El más común de este tipo de cánceres es el osteosarcoma. No obstante se ha asociado al retinoblastoma con cánceres de ovario , mama, pulmón, próstata y vejiga en otras etapas de la vida. Aún no se conoce la causa del cáncer secundario.
Referencias
1. Donaldson SS, Egbert PR, lee WH: retinoblastoma. In: Pizzo PA, Poplack DG: Principles and practices of Pediatric Oncology. 2nd ed. Philadelphia ,PA : JB Lippincott, 1993 , pp 683-696. [ Links ]
2. Shields Cl, Shields JA, Shah P: retinoblastoma in older children. Ophthalmology 98: 395-9, 1991. [ Links ]
3.Zajaczek S, Jakubowska A, Kurzawski G, el al.: Age at diagnosis to discriminate those patients for whim constitutional DNA sequencing is appropriate in sporadic unilateral retinoblastoma. Eur J Cancer 43: 1919-21, 1998.
4.Herzog S, Lohmann DR, BuitingK, et al : Marked differences in unilateral isolated retinoblastomas from young and older children studied by comparative genomic hybridization . Hum Genet 108: 98-104, 2001 [ Links ]
5.Abramson DH, Mendelsohn ME, servodidio CA, et al.: Familial retinoblastoma: where and when? Acta Ophthalmol Scand 76: 334-8, 1998. [ Links ]
6.Agramson DH, Frank CM: Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. Ophthalmology 105 :573-9; Discussion 579-80, 1998. [ Links ]
7.Paulino AC: Trilateral retinoblastoma: Is the location of the intracranial tumor important? Cancer 86 (1) : 135-41,1999. [ Links ]
8.Kivela T: Trilateral retinoblastoma: A meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17: 1829-37, 1999. [ Links ]
9.Nunier F, Peseia G, Jotterand Bellomo M, el.al: Evidence of somatic and ferminal mosaicism in pseudo-Iow penetrant hereditary retinoblastoma, by constitutional and single-sperm mutation análisis. Am J Hum Genet 63 (6): 1903-8, 1998.
10. Munier F, Pescia G, Jotterand - Bellomo M, et. al : constitutional karyotype in retinoblastoma. Case report and review of literature. Ophthalmic paediatr Genet 10 : 129-50, 1989. [ Links ]
11.Koperman JE, Mc lean IW, Rosenberg SH: multivariate analysis of risk factors for metastasis in retinoblastoma treated by enucleation Ophthalmology 94: 371-7,1987. [ Links ]
12.Schwimer CJ, Prayson RA: Clinicopathologic study of retinoblastoma including MIB-1, p53, and CD99 inmunohistochemistry. Ann Diagn Pathol 5: 148-54, 2001. [ Links ]
13.Howarth C, Meyer D, Hustu HO, et.al.: Stagerelated combined modality treatment of retinoblastoma. Results of a prospective study. Cancer 45: 851-8, 1980. [ Links ]
14.Messmer EP, Heinrich T, et.al: Risk factors for metastases in patients with retinoblastoma. Ophthamology 98 : 136-41, 1991. [ Links ]
15.Friedman Dl, Himelstein B, Shields Cl, et.al: Chemoreduction and local ophthalmic therapy for intraocular retinoblastoma. J Clin Oncol 18: 127,2000. [ Links ]
16. Shields Cl, Santos MC, Diniz W, et al.: Thermotherapy for retinoblastoma. Arch Ophthalmol 117 : 885-93, 1993. [ Links ]
17.Shields Cl, Shields JA, Cater J, et .al: Plaque radiotherapy for retinoblastoma: long term tumor control and treatment complications in 208 tumors.Ophthalmology 108 : 2116-21, 2001. [ Links ]
18.Geindeiz K, Shields Cl; Shields JA, et . al. : The outcome of chemoreduction treatment in patients with risk-ellsworth group V retinoblastoma. Arch Ophthalmol116: 1613-7, 1998. [ Links ]
19.Rootman J, Hofbauer J, ElIsworth RM, et.al: Invasion of the optic nerve by retinoblastoma: a clinicalpathological study. Can J Ophthalmol 11 :106-14,1976. [ Links ]
