










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina Legal de Costa Rica
On-line version ISSN 2215-5287Print version ISSN 1409-0015
Med. leg. Costa Rica vol.32 n.2 Heredia Sep./Dec. 2015
Reporte de caso
Hemangioblastoma cereberoloso en el síndrome de von Hippel-Lindau, como causa de muerte. Reporte de un caso
Kennette Villalobos León*+
Resumen:
El síndrome de von Hippel-Lindau es una enfermedad caracterizada por el desarrollo de tumores como hemangioblastomas del sistema nervioso central y de la retina, quistes renales, hepáticos y pancreáticos; carcinoma renal de células claras, feocromocitoma, así como adenomas en el oído interno, la nariz y la laringe. En la mayoría de los casos hay un antecedente familiar positivo del síndrome 8. A continuación se presenta el caso de una femenina de 35 años de edad, sin antecedentes personales patológicos conocidos, con antecedente familiar positivo por la enfermedad de von Hippel-Lindau. Según la historia de sus familiares, la mujer presentaba ataques
de rigidez de dos semanas de evolución, posterior a uno de estos ataques fue atendida por paramédicos quienes la declararon fallecida. Según la autopsia médico legal se diagnosticó una hidrocefalia obstructiva secundaria a hemangioma cerebeloso y hallazgos compatibles con el Síndrome de von Hippel-Lindau. El objetivo de este artículo es resaltar los hallazgos postmortem de esta entidad y su relación con la causa de la muerte.
Palabras clave:
Von Hippel-Lindau, autosómico dominante, feocromocitoma, hemangioblastoma.
Abstract:
Von Hippel-Lindau syndrome is characterized by tumors as central nervous system and retinal hemangioblastomas; renal, liver and pancreas cysts; clear cells renal carcinoma, pheochromocytoma and adenomas of the ear, nose and larynx. In most cases there is a positive family history of the syndrome 8. The follow case is about a female how was 35 years old without known medical history. She had a positive family history of von Hippel-Lindau, and her family told us about she had stiffness attacks and two weeks later had another stiffness attack so it was treated by paramedics who declared her dead. In agreement with the forensic autopsy findings the diagnosis was hydrocephalus induced by brain stem hemangioma and the findings were compatible with von Hippel-Lindau. The aim of this article is to highlight the most important postmortem findings of this syndrome and its relation to the cause of death.
Keywords:
Non traumatic spleen rupture, surgical instrumentation, multiple transfusions, bleeding on massive transfusions.
Introducción
El síndrome de Von Hippel- Lindau (VHL) es una entidad poco frecuente, cuya sintomatología es variada y su diagnóstico temprano premortem no es fácil8. Este síndrome se caracteriza por asociar una gran variedad de tumores, tanto de tipo maligno como benigno, en distintas partes del cuerpo11. Las alteraciones patológicas más frecuentes son los hemangioblastomas del sistema nervioso central, los angiomas retinianos, los tumores renales de células claras y los feocromocitomas 14. No obstante, también se ha relacionado la presencia de tumores del oído, la nariz y la garganta. La presencia de hemangioblastomas a nivel del sistema nervioso central debe hacer sospechar la presencia de VHL, principalmente si son múltiples 12.
Muchos de los pacientes con esta enfermedad pueden cursar asintomáticos, sin embargo, pueden presentar síntomas relacionados con la ubicación de los tumores desarrollados 8.
Presentación de caso
Femenina caucásica de 35 años de edad, con sobrepeso (IMC 28,72), sin antecedentes personales patológicos conocidos, con antecedente familiar positivo por la enfermedad de von Hippel-Lindau. Según la declaración de sus familiares, la mujer presentaba "ataques de rigidez" de dos semanas de evolución y posterior a uno de estos ataques fue atendida por paramédicos quienes la declararon fallecida.
Al examen externo, durante la autopsia médico legal, se evidenció congestión cervicofacial y cianosis distal en las extremidades superiores.
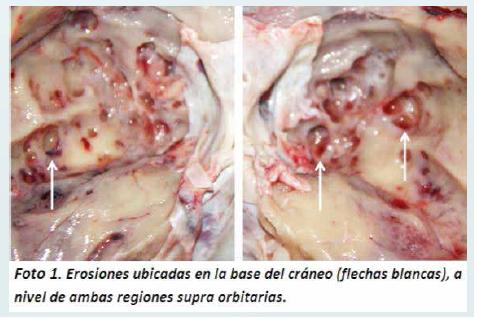
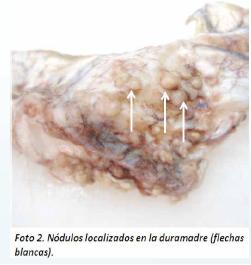
Al examen interno del cráneo se observó erosiones en el hueso frontal a nivel supra orbitario de hasta 0.5 cm de diámetro (Foto 1), varios nódulos en la duramadre de color amarillento, de consistencia duroelástica, adheridos a la cara ósea a nivel supra orbitario de hasta 0.5 cm de diámetro, cuya ubicación coincidía con las erosiones supra citadas (Foto 2).
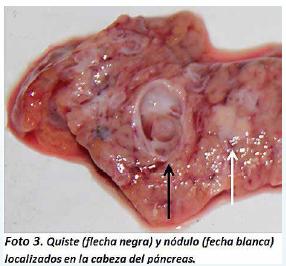
El cerebro pesó 1276 gr con borramiento de surcos y aplanamiento de circunvoluciones. Posteriormente el cadáver fue abierto con la técnica de Rokitansky modificada y se documentó a nivel de la pleura visceral petequias y hemorragias subpleurales distribuidas de manera difusa en ambos pulmones, los cuales eran de consistencia hulosa a la palpación y al corte resumaban líquido rosado espumoso. A nivel del abdomen, el páncreas pesó 75 gr y al corte tenía múltiples quistes de entre 0.5 y 1 cm de diámetro localizados en toda su extensión. Además presentó en la cabeza del páncreas un nódulo de 1 cm de diámetro de color blanco-amarillento y de consistencia sólida (Foto 3).
Al corte del riñón derecho presentó una masa localizada en el polo superior, redondeada de 2 cm longitud x 2 cm de ancho x 2 cm de espesor, de color amarillo con áreas de hemorragia sin aparente infiltración de la cápsula.
El estudio microscópico de los nódulos descritos en la duramadre evidenció fragmentos de tejido cerebral con neuronas y células gliales rodeadas de tejido fibroso. Hubo protrusión focal de la capa molecular del cerebro con neuronas y glía desordenadas en el corte de corteza frontal y vacuolación de la neuropila subependimaria con congestión intravascular en el corte de núcleos basales.
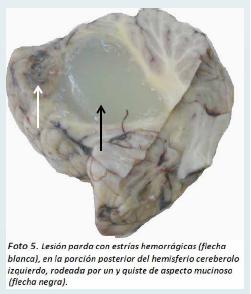
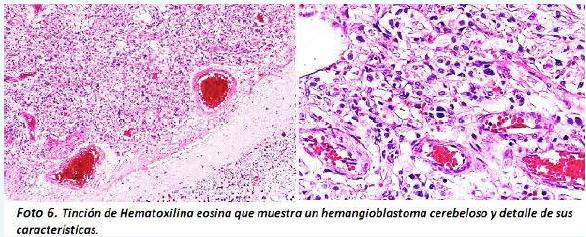
En el cerebelo se identificó una neoplasia con dos poblaciones de células conformadas por una fina malla de capilares con células endoteliales hipertrofiadas y por células intersticiales, la mayoría de citoplasma claro vacuolado, con moderado pleomorfismo nuclear (Foto 6).
Se observó gliosis reactiva con fibras de Rosenthal y cuerpos granulares en la periferia, además de una gran cantidad de material amorfo acelular eosinofílico correspondiente con el contenido del quiste descrito macroscópicamente.
No se encontró necrosis neuronal, infiltrados inflamatorios o hemorragias.
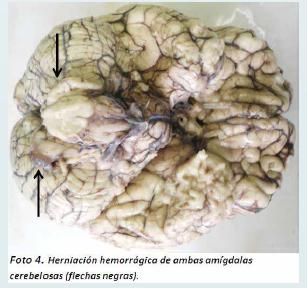
Estos hallazgos permitieron hacer el diagnóstico de un hemangioblastoma del vermis cerebeloso grado I con compresión extrínseca del acueducto cerebral y edema cerebral severo con hernia de ambos uncus del hipocampo y de ambas amígdalas cerebelosas. Además se demostró heterotopia glioneuronal leptomeningea focal y parameningea.
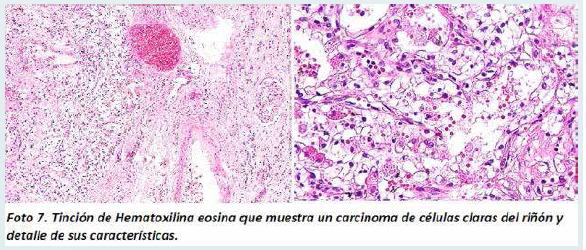
El estudio histopatológico mostró a nivel del riñón una neoplasia epitelial conformada por células aumentadas de tamaño con abundante citoplasma claro, con núcleos pequeños y nucléolo inconspicuo. Estas células estaban dispuestas en túbulos y nidos, con una delicada red vascular, con áreas de hemorragia y macrófagos con hemosiderina, hallazgos que corresponden a un carcinoma de células claras del riñón (Foto 7) con Micrometástasis pulmonares.
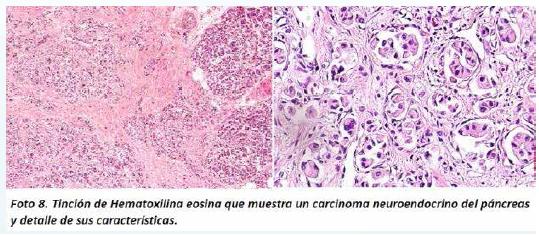
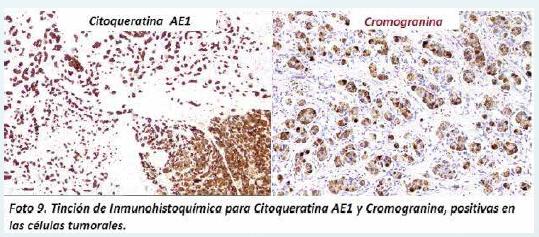
A nivel del páncreas se encontró una neoplasia de células epiteliales, con núcleo de tamaño moderado, con cromatina en sal y pimienta, algunas con núcleos hipercromáticos aumentados de tamaño y pleomórficos. Estas células dispuestas en túbulos y nidos, algunos con patrón infiltrativo y con canibalismo celular (Foto 8). El estudio de inmunohistoquímica demostró que estas células neoplásicas eran positivas para citoqueratina AE1, sinaptoficina y cromogranina, lo cual permitió hacer el diagnóstico de un carcinoma neuroendocrino bien diferenciado del páncreas (Foto 9).
También se observaron microquistes revestidos por células cúbicas, de núcleo redondeado con moderado citoplasma eosinofílico que correspondieron a cistoadenomas serosos de páncreas.
El miocardio mostró áreas eosinofílicas y fragmentación de fibras con presencia de estriaciones que se interpretó como cambios isquémicos recientes.
En cuanto a los resultados de exámenes auxiliares el análisis de determinación de alcohol y drogas de abuso en sangre periférica fueron negativos.
Según lo anterior, se concluyó que la causa de muerte fue debida a un síndrome de von Hippel-Lindau con un hemangioblastoma del vermis cerebeloso que produjo edema cerebral severo y hernia de amígdalas cerebelosas, siendo la manera de muerte natural desde el punto de vista médico legal.
Discusión
Cerca del 70% de los pacientes portadores de VHL tienen carcinomas renales bilaterales y/o quistes múltiples renales, y su aparición es más común luego de los 20 años 8. El tumor de células renales es el tumor más frecuente en el riñón adulto y el subtipo más frecuente es el de células claras, cuya morfología típica es la de un tumor de consistencia sólida con componente quístico y una cápsula fibrosa en la periferia. Los tumores pancreáticos neuroendocrinos pueden aparecer de manera esporádica o estar ligados a enfermedad tal como lo es VHL 2.
Los feocromocitomas son paragangliomas simpáticos de ubicación adrenal, estos tumores son capaces de producir, secretar y metabolizar catecolaminas 1. Son importantes en la clasificación del tipo de VHL. Comúnmente se desarrollan en adultos jóvenes y pueden estar en ambas glándulas adrenales. Aproximadamente 35% de los pacientes con feocromocitoma son asintomáticos, sin embargo los síntomas típicos por catecolaminas son: taquicardia, diaforesis, hipotensión postural, taquipnea, piel fría y pegajosa, cefalea, dolor precordial, palpitaciones, nauseas, vómitos o dolor epigástrico. Los pacientes pueden presentar catecolaminas elevadas en sangre, sin embargo es más utilizado
la presencia de ácido vainillilmandélico en orina como método diagnóstico 14. Generalmente estos tumores se encuentran bien delimitados y hasta encapsulados, comúnmente son pequeños (3-5 cm) de color gris, con importante vascularización. Debido a que se trata de un tumor benigno, el único criterio confiable de malignidad es la metástasis 1.
Otro tipo de tumores neuroendocrinos que se han relacionado con VHL, a pesar de su poca frecuencia, son los paragangliomas parasimpáticos, que consisten en tumores no secretores de catecolaminas, localizados en la cabeza y el cuello, y que según algunos autores, deben ser considerados dentro del espectro diagnóstico para esta enfermedad4.
Conclusión
El síndrome de von Hippel- Lindau es una entidad cuya sintomatología puede ser variada y poco específica, siendo más súbita cuando hay afección del sistema nervioso central. El diagnóstico postmortem no escapa de esta complejidad, por lo que requiere de un cuidadoso examen de los órganos y la descripción detallada de todos los hallazgos, así como el apoyo en la histología y neuropatología como medios diagnósticos indispensables en este tipo de casos.
Partiendo del hecho de que solamente una quinta parte de los casos de esta enfermedad se producen de manera esporádica es recomendable que el médico informe a los familiares de la persona diagnosticada con VHL para que estén al tanto de cualquier hallazgo clínico sugestivo que requiera atención médica para el diagnóstico y/o tratamiento pertinente.
Bibliografía
1. Rojas, E. I. (2012). Feocromocitoma. Revista Médica de Costa Rica y Centroamérica, LXIX, 69-72. [ Links ]
2. Weisbrod, A. B., Kitano, M., Thomas, F., Williams, D., Gulati, N. & Gesuwan, K., et. al. (2014, Feb.). Assessment of Tumor Growth in Pancreatic Neuroendocrine Tumors in von Hippel Lindau Syndrome. J Am Coll Surg, 218(2), 163-169. [ Links ]
3. Frantzen, C., Kruizinga, R. C., van Asselt, S. J., Zonnenberg, B. A., Lenders, J. W. & de Herder, W. W., et al. (2012). Pregnancy-related hemangioblastoma progression and complications in von Hippel-Lindau disease. Neurology, 79(8), 793-796. [ Links ]
4. Gaal, J., van Nederveen, F. H., Erlic, Z., Korpershoek, E., Oldenburg, R. & Boedeker, C. C. (2009, Nov.). Parasympathetic Paragangliomas Are Part of the Von Hippel-Lindau Syndrome. J Clin Endocrinol Metab, 94(11), 4367-4371. [ Links ]
5. García, S., Pérez, C., & Cacho, G. (2011). Alteraciones del Líquido cefalorraquídeo y su circulación: hidrocefalia, pesudotumor cerebral y síndrome de presión baja. Medicine, 4814-4824. [ Links ]
6. Hoffmanna , J. & Goadsbyb, P. J. (2013). Update on intracranial hypertension and hypotension. Current Opinion, 26, 240-247. [ Links ]
7. Jagannathan, J., Lonser, R., Smith, R., DeVroom, H. L. & Oldfield, E. H. (2008, Feb.). Surgical management of cerebellar hemangioblastomas in patients with von Hippel–Lindau disease. J Neurosurg, 108(2), 210-222. [ Links ]
8. O Brien, F. J., Danapal, M., Jairam, S. & Lala, A. K. (2014). Manifestations of Von Hippel Lindau syndrome: A retrospective national review. Q J Med, 291-296. [ Links ]
9. Pavesi, G., Berlucchi, S., Munari, M., Manara, R., Scienza, R. & Opocher, G. (2010). Clinical and surgical features of lower brain stem hemangioblastomas in von Hippel-Lindau disease. Acta Neurochir, 287-292. [ Links ]
10. Peyre, M., David, P., Van Effenterre, R., Francois, P., Thys, M. & Emery, E., et. al. (2010, Sep.). Natural History of Supratentorial Hemangioblastomas in von Hippel-Lindau Disease. Neurosurgery, 67, 577-587. [ Links ]
11. Rao, P., Monzon, F., Jonasch, E., Matin, S. & Tamboli, P. (2014). Clear cell papillary renal cell carcinoma in patients with von Hippel- Lindau syndromeclinicopathological features and comparative genomic analysis of 3 cases. Human Pathology, 1966-1972. [ Links ]
12. Schief, D. & Patrick, B. (2005). Principles of Neuro-Oncology.
13. Sharma, P., Singh, V.,
14. Vaganovs, P., Bokums, K., Miklasevics, E., Plonis, L. Zarina, L. & Geldners,
* Médico Residente Departamento de Medicina Legal,


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}