










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina Legal de Costa Rica
On-line version ISSN 2215-5287Print version ISSN 1409-0015
Med. leg. Costa Rica vol.32 n.1 Heredia Jan./Mar. 2015
Revisión bibliográfica
Productos finales de glicación (AGES) y la nefropatía diabética
Carlos Carvajal Carvajal*+
Resumen:
Los productos finales de glicación (AGEs) son un grupo heterogéneo de moléculas generadas por medio de reacciones no enzimáticas de glicación y de oxidación de proteínas, lípidos y ácidos nucleicos. La formación aumentada de AGEs ocurre en condiciones tales como la diabetes mellitus y el envejecimiento. AGEs median sus efectos a través de tres mecanismos principales: 1) entrecruzamiento con proteínas de la matriz extracelular, afectando las propiedades mecánicas de los tejidos, 2) entrecruzamiento con proteínas intracelulares alterando sus funciones fisiológicas y 3) unión a sus receptores de superficie RAGE para inducir múltiples cascadas
de señales intracelulares.
La acumulación de AGEs en las proteínas tisulares ha sido implicada en las complicaciones vasculares diabéticas, tales como la retinopatía, la nefropatía y la neuropatía. En la nefropatía diabética los AGEs contribuyen al desarrollo y progresión de esta enfermedad renal.
Palabras claves:
Productos finales de glicosilación avanzada, Reacción de Maillard, Complicaciones de la diabetes, Membrana basal glomerular.
Abstract:
Advanced glycation end products (AGEs) are a heterogenous group of molecules that are generated through nonenzimatic glycation and oxidation of proteins, lipids and nucleic acids. Enhanced formation and accumulation of AGEs has been reported to occur in conditions such as diabetes mellitus as well as in natural aging. AGEs mediate their effects through three main mechanism: 1) cross linking extracellular (matrix) proteins thereby affecting tissue mechanical properties, 2) cross linking intracellular proteins thus altering their physiological functios and 3) binding to their cell surface receptor RAGE to inducing multiple intracellular signalling cascades.
The accumulation of AGEs in tissue proteins has been implicated in diabetic vascular complications, such as retinopathy, nephropathy and neuropathy. In the diabetic nephropathy AGEs contribute to the development and progression of this renal disease.
Key words:
Glycosylation end products, advanced; Maillard reaction; Diabetes complications; Glomerular basement membrane.
Origen y definición de los productos finales de glicación (ages)
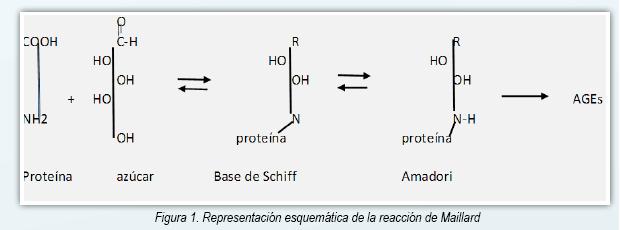
Los AGE (advanced glycation end products) son un espectro de compuestos heterogéneos que derivan de proteínas, lípidos y ácidos nucleicos que son glicados y oxidados en forma no enzimática en un proceso llamado reacción de Maillard (1, 2). La glucosa tiene un papel primordial en el proceso debido a su alta concentración en el plasma, aunque otros azúcares reductores son implicados también (fructosa, galactosa, manosa y xilulosa) (3). La reacción de Maillard se inicia como una reacción entre el grupo carbonilo de un azúcar reductor y el grupo amino libre de una proteína, de un lípido o de un ácido nucleico y lleva a la formación de una Base de Schiff inestable. Esta reacción es reversible y requiere de pocas horas para ocurrir (4). A través de varias semanas estos compuestos lábiles originan un producto Amadori más estable. Posteriormente y en plazo de meses a años una pequeña parte de los compuestos
Amadori sufre otras reacciones irreversibles (oxidación, deshidratación y degradación) originando los AGEs, que son compuestos altamente estables (Figura 1) (5,6). En las proteínas los AGE se forman sobre residuos de lisina o arginina predominantemente (7). Además de la reacción de Maillard otras vías pueden originar los AGE, como por ejemplo la autooxidación de la glucosa y la peroxidación de los lípidos que originan derivados dicarbonílicos a partir de un incremento del estrés oxidativo (8). Figura 1
Las especies dicarbonílicas derivadas de la glucosa, tal como el metilglioxal y el glioxal, son altamente reactivas y se cree que son los intermediarios metabólicos precursores de la mayoría de los productos glicados y no directamente la glucosa, que es un compuesto poco reactivo. Como estos compuestos se producen intracelularmente hay una mayor posibilidad de glicar las proteínas intracelulares que las extracelulares. En consecuencia los niveles de los AGE tienden a ser mayores en las proteínas celulares que en las plasmáticas (9).
La glicación es el término más general para la unión no enzimática de un azúcar a otra biomolécula. La glicación es un proceso no enzimático y la glicosilación es el proceso enzimático formando un enlace glicosídico (10).
Los AGE son producidos normalmente y se acumulan con la edad. En el envejecimiento normal la formación de AGE es más lenta y ocurre particularmente sobre proteínas de larga vida. En la diabetes la formación y acumulación de los AGEs se acelera debido a los altos niveles de glucosa sanguínea. En diversos estudios el nivel sérico de los AGE es mayor en los diabéticos que en su contraparte no diabética (2,9).
Diversos estudios han asociado los AGE séricos y tisulares con las complicaciones micro y macrovasculares de la diabetes (aterosclerosis, retinopatía, nefropatía y neuropatía) y con otras patologías, entre ellas la enfermedad de Alzheimer (2, 5,10-14).
Los RAGEs son receptores multi-ligandos que median muchos de los efectos de los AGEs y son expresados en muchos tejidos a nivel de superficie celular de células endoteliales, fagocitos mononucleares, monocitos, macrófagos, hepatocitos microglia, células de músculo liso, astrocitos, ciertas neuronas, células mesangiales y podocitos entre otras. Bajo condiciones normales la expresión de los RAGEs es baja, mientras que en condiciones patológicas, tales como la inflamación y la diabetes, hay un mayor nivel de los mismos, coincidente con un mayor nivel de AGE (2).
AGE y nefropatía
La nefropatía diabética (ND) es la principal causa de la enfermedad renal terminal en el mundo y una de las principales causas de mortalidad en los pacientes diabéticos (15). La ND se caracteriza por un incremento progresivo en los niveles de albuminuria, hipertensión, glomeruloesclerosis y una eventual reducción de la tasa de filtración glomerular (GFR).
La ND temprana puede definirse como una microalbuminuria persistente medida en dos ocasiones con una tasa de excreción de 20 a 200 ug/min ó 30 a 300 mg/24 h. La ND franca se define como una albuminuria más allá del estado de microalbuminuria. Los pacientes con diabetes tipo 1 y 2 presentan las mismas características en la ND (16).
Un elemento clave en la ND es la serie de cambios que ocurren en la estructura de filtración glomerular y se considera a la hiperglicemia como un factor principal en la iniciación y progresión de esta patología (16).
La barrera de filtración glomerular consta de tres capas: una capa de células endoteliales glomerulares, la membrana basal glomerular (MBG) y una capa de células especializadas llamadas podocitos. Hay además células mesangiales que proveen apoyo estructural a los capilares. El endotelio es altamente fenestrado y sintetiza la MBG y el glucocalix.
Esta última estructura es una capa de proteoglucanos, glicosaminoglicanos (GAG), glucolípidos y proteínas plasmáticas atrapadas. Los proteoglucanos se distribuyan de tal forma que obstruyen parcialmente las fenestraciones endoteliales y junto con los GAG proveen cargas negativas que constituyen una barrera contra la filtración de la albúmina y de otras proteínas. La MBG está formada principalmente de colágeno tipo IV, proteoglucanos y laminina. El colágeno y la laminina brindan soporte a la pared capilar y los proteoglucanos con sus cargas negativas proveen a la MBG de su selectividad de carga. Los podocitos forman extensiones tipo tentáculo que les permiten cubrir toda la superficie de filtración, se hallan cubiertos por el glucocálix y se adhieren a la MBG a través de uniones entre proteínas (17).
El paso a través de la barrera de filtración para proteínas de bajo peso molecular (PM) (≤ 30 KDa y radio < 20 Å) es casi totalmente libre, para proteínas de PM intermedio, como la albúmina, solo se filtra una pequeña fracción y para proteínas de alto PM virtualmente no hay filtración (18).
En la enfermedad renal progresiva se considera que la activación endotelial y la pérdida de la selectividad por carga constituyen los primeros eventos (17). La activación endotelial lleva a la expresión de moléculas de adhesión, como ICAM y VCAM, que son necesarias para la adhesión de leucocitos (19-21). Además el endotelio activado expresa también factores quimiotácticos tales como MCP-1 y otras citokinas proinflamatorias como TNF-β. La expresión endotelial de estos factores contribuye al desarrollo de la inflamación, condición necesaria para el desarrollo de la ND.
Adicionalmente los macrófagos activados producen localmente una serie de factores proinflamatorios, profibróticos y antiangiogénicos que incluyen TNF-α, IL-1, IL-6, especies reactivas de oxígeno (ROS), PDGF, TGF-β, angiotensina II (ATII) y endotelina. El factor TNF-α está implicado también en la producción local de ROS, en el aumento de la permeabilidad a la albúmina, en el reclutamiento de monocitos y en la disminución de la GFR (21). Los ROS contribuyen al daño renal en varias formas: induciendo la disfunción endotelial renal y la microalbuminuria, la acumulación de matriz, la expansión mesangial y la fibrosis (22). El infiltrado de macrófagos se asocia a una inflamación crónica de bajo grado. Los macrófagos pueden interactuar con las células renales para generar un microambiente proinflamatorio que amplifica el daño y promueve la fibrosis.
La acumulación de AGEs en el riñón puede contribuir a la alteración progresiva de la arquitectura renal y la pérdida de la función renal en los pacientes por dos mecanismos: glicación de componentes de la barrera de filtración glomerular o adyacentes a la misma y activación de vías intracelulares por medio de la interacción AGE: RAGE.
Glicación de componentes.
La glicación del colágeno tipo IV y de laminina reduce su capacidad para interactuar con los proteoglicanos incrementando la permeabilidad vascular a la albúmina y la glicación de las proteínas de la matriz aumenta su resistencia a las proteasas contribuyendo al engrosamiento de la MBG y a la expansión mesangial (7,23).
Interacción AGE: RAGE
La diabetes causa una mayor producción y acumulación de AGE y los AGE y/o la condición diabética aumentan la expresión de RAGEs en varios tipos celulares. Los pacientes con enfermedad renal terminal tienen dos veces más AGE en los tejidos que los pacientes diabéticos con enfermedad renal (24).
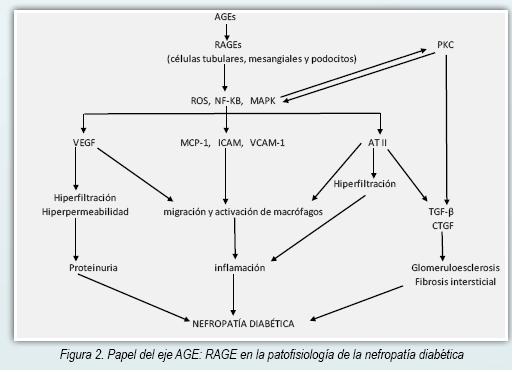
La interacción de AGE: RAGE activa la enzima NADPH oxidasa causando un incremento de ROS y generando estrés oxidativo (25,26). Los ROS son citotóxicos a nivel renal y a través de la activación de la vía de la MAPK (MAP kinasas), NF-kB y de la Proteína Kinasa C (PKC) en las células mesangiales y túbulointersticiales promueven reacciones inflamatorias y fibrogénicas por medio de la producción aumentada de VEGF, TGF-β y CTGF (20,27). TGF-β no solo estimula la síntesis de la matriz sino que también inhibe su degradación, estando involucrada en la esclerosis glomerular (28). En ratones transgénicos que sobreexpresan TGF-β se observa disfunción renal caracterizada por proteinuria, glomeruloesclerosis y fibrosis túbulointersticial (29). En la Figura 2 puede verse el proceso patofisiológico que lleva al daño renal a partir del eje AGE: RAGE.
La interacción AGE: RAGE induce la producción de MCP-1 (30) propiciando un infiltrado mononuclear a nivel renal como parte de una reacción inflamatoria crónica (31) y también promueve la inducción de apoptosis de las células mesangiales y contribuye por esa vía a la hiperfiltración glomerular (15). Figura 2
El papel de la asociación AGE: RAGE se ha podido dilucidar utilizando ratones transgénicos. La cepa OVE26 de ratones diabéticos presenta albuminuria, esclerosis glomerular, alteración de los podocitos, engrosamiento de la MBG y disminución franca de la GFR. Al alterar dicha cepa para que no exprese RAGE se nota una disminución significativa de todas las alteraciones citadas (32). Otros estudios con cepas de ratones transgénicos con sobreproducción de RAGE resultaban en hipertrofia glomerular, expansión mesangial difusa, infiltración celular inflamatoria y fibrosis intersticial al compararlos con ratones sin sobreexpresión de RAGE (33).
La hiperglicemia estimula la producción de Angiotensina II (AT II) y ésta última incrementa la producción de ROS en las células renales a través de la activación de la NADPH oxidasa y en última instancia la producción de AGEs en el riñón diabético (20, 34). En este sentido se puede hablar de un eje AGE-RAGE-ATII. Además, la producción aumentada de ATII causaría la hipertensión observada en los pacientes con ND (35).
Conclusiones
Los AGEs son un grupo heterogéneo de moléculas originadas por la glicación y oxidación de proteínas, lípidos y ácidos nucleicos.
Los AGEs se forman normalmente a través de varios pasos químicos a corto y largo plazo. El proceso modifica lípidos, proteínas y ADN y su formación y acumulación se favorece con la hiperglicemia y el envejecimiento.
La acumulación de AGE se ha asociado a diversas patologías observadas en los pacientes diabéticos a largo plazo (retinopatía, neuropatía, nefropatía y aterosclerosis)
Los AGE actúan por varios mecanismos entre ellos la glicación de biomoléculas y por medio de su receptor RAGE activando numerosas vías intracelulares.
Referencias
1. Frye, E. B., Degenhardt, T. P., Thorpe, S. R. & Baynes, J. W. (1998). Role of Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J Biol Chem, 273, 18714-18719. [ Links ]
2. Hegab, Z., Gibbons, S., Neyses, L. & Mamas, M. (2012). Role of advanced glycation end products in cardiovascular disease. World J Cardiology, 4, 4, 90-102. [ Links ]
3. Piarulli, F., Sartore, G. & Lapolla, A. (2013). Glyco-oxidation and cardiovascular complications in type 2 diabetes: a clinical update. Acta Diabetol, 50, 101- 110. [ Links ]
4. Watkins, N. G., Thorpe, S. R. & Baynes, J. W. (1985). Glycation of amino groups in protein. J Biol Chem, 260, 10629-10636. [ Links ]
5. Schalkwijk, C. & Miyata, T. (2012). Early- and advanced non-enzimatic glycation in diabetic vascular complications: the search for therapeutics. Amino Acids, 42, 1193-1204. [ Links ]
6. Taguchi, A., Blood, D. C., Del Toro, G. & Canet, A., et al. (2000). Blockade of RAGE-amphoterin signaling suppresses tomour growth and metastases. Nature, 405, 354-360. [ Links ]
7. Ramasamy, R., Yan, S. & Schmidt, A. (2012). Advanced glycation endproducts: from precursors to RAGE: round and round we go. Amino Acids, 42, 4, 1151-1161. [ Links ]
8. Luevano, C. & Chapman, K. (2010). Dietary advanced glycation end producti and aging. Nutrients, 2, 1247-1265. [ Links ]
9. Boon, P., Pun. L. & Murphy, M. (2012). Pathological significance of mitochondrial glycation. International J Cell Biology, 2012, 1-13. [ Links ]
10. Zhang, Q.,
Res, 8, 2, 754-769.
11. Bornfeldt, K. & Tabas,
12. Yan, S., Chen, D., Yan S., Guo, L. & Chen, J. (2013). RAGE is a key cellular target for Aβ-induced perturbation in Alzheimer´disease, Front Biosci (Schol Ed), 4, 240-250. [ Links ]
13. Li, X. H., Cheng, X. S., Jiang, X., Zhang, Y., Lv, B. L., Liu, R., et al. (2013). Glycation exacerbates the neuronal toxicity of β-amyloid. Cell Death and Disease, 4, 1-110
14. Deane, R., Singh, I., Sagare, A.,
15. Yamagishi, S. I. & Matsui, T. (2010). Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxidative medicine and cellular longevity, 3, 2, 101-108. [ Links ]
16. Kolset, S., Reinholt, F. & Jenssen, T. (2012). Diabetic nephropathy and extracellular matrix. J Histochemistry & Cytochemistry, 60, 12, 976-986. [ Links ]
17. Garg, P. & Rabelink, T. (2011). Glomerular proteinuria: a complex interplay between unique players. Adv Chronic Kidney, 18, 4, 233-242. [ Links ]
18. Vinge, L., Lees, G., Nielsen, R., Clifford E., et al. (2010). The effect of progressive glomerular disease on megalin-mediated endocytosis in the kidney. Nephrol Dial Transplant, 25, 2458-2467. [ Links ]
19. Ramasamy, R., Yan, S. & Schmidt, A. (2012). The diverse ligand repertoire of the receptor advanced glycation endproducts & pathways to the complications of diabetes. Vascul Pharmacol, 57, 5-6, 160-167. [ Links ]
20. Tabit, C. (2012). Endothelial dysfunction in diabetes mellitus: molecular mechanism and clinic implications. Rev Endocr Metab Disord, 11, 1, 61-74. [ Links ]
21. Lim, A. & Tesch, G. (2012). Inflamation in diabetic nephropathy. Mediators of inflammation, 2012, 1-12. [ Links ]
22. Raimundo, M. & López, J. (2011). Metabolic Syndrome, chronic kidney disease and cardiovascular disease: a dynamic and life-threatening triad. Cardiology Research ad Practice, 2011, 1-16. [ Links ]
23. Brownlee, M. & Lecture, L. (1994). Glycation and diabetic complications. Diabetes, 43, 836-841. [ Links ]
24. Makita, Z., Radoff, S., Rayfield, E. J., Yang, Z., et al. (1991). Advanced glycosylation end products in patients with diabetic nephropathy.
25. Gella, A. & Durany, N. (2009). Oxidative stress in Alzheimer disease. Cell Adhesion & Migration, 3, 1, 88-93. [ Links ]
26. Younessi, P. & Yoonessi, A. (2011). Advanced glycation end-products and products and their receptor-mediated roles: inflammation and oxidative stress.
27. Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature, 414, 813-820. [ Links ]
28. Border, W. A. & Noble, N. A. (1994). Transforming growth factor beta in tissue fibrosis.
29. Goldfarb, S. & Ziyadeh, F. (2001). TGF-β: a crucial component of the pathogenesis of diabetic nephropathy. Transac Am Clin Climatol Assoc, 112, 27-33. [ Links ]
30. Panee, J. (2012). Monocyte chemoattractant protein (MCP-1) in obesity and diabetes. Cytokine, 60, 1, 1-12. [ Links ]
31. Yamagishi, S., Inagaki, Y. & Okamoto, T. (2002). Amano S. Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattracant protein-1 in human mesangial cells. J Biol Chem, 277, 20309-20315. [ Links ]
32. Reiniger, N., Lau, K., McCalla, D., Eby, B., Cheng, B., Lu, Y., et al. (2010). Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes, 59, 2043-2055. [ Links ]
33. I nagi, R., Yamamoto, Y., Nangaku, M. & Usuda, N. (2006). A severe diabetic nephropathy model with early development of nodule-like lesions induced by megsin overexpression in RAGE/iNOS transgenic mice. Diabetes, 55, 356- 366. [ Links ]
34. Forbes, J. M., Cooper, M. E., Thallas, V. & Burns, W. C. (2002). Reduction of the accumulation of advanced glycation end products by ACE inhibition in experimental diabetic nephropathy. Diabetes, 51, 3274-3282. [ Links ]
35. Kanwar, Y. S., Sun, L., Xie, P. & Liu, F. Y. (2011). A glimpse of various pathogenetic mechanism of diabetic nephropathy. Annu Rev Patol, 6, 395- 423. [ Links ]
* Microbiólogo. Especialista en Química Clínica. E-mail: ccarvajal313@yahoo.com
Recibido para publicación el 12 de agosto de 2014 Aceptado el 16 de setiembre de 2014


{kind=link}
{kind=link}