









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Fragaria x ananassa es el nombre con el que se conoce a todas las variedades de fresa que se cultivan mayoritariamente en la actualidad (1). México es el tercer proveedor de fresa fresca al mercado internacional; según datos de la Secretaría de Agricultura, Ganadería, Desarrollo Rural, Pesca y Alimentación (2) en el año 2016 se produjeron 468.25 miles de toneladas de la fruta en el país, y se estima que para el 2030 la producción generaría un ingreso de 866.71 millones de dólares.
Numerosos programas de mejora de este cultivo se han dirigido a optimizar las características del fruto, la resistencia a patógenos y adversidades climáticas (3). No obstante, la biotecnología se ha planteado objetivos más específicos para potenciar su efecto beneficioso sobre la salud humana, debido al contenido de vitaminas, minerales y antioxidantes de la fruta (4), además de ser fuente de metabolitos secundarios como las antocianinas, ácidos fenólicos, taninos y flavonoides (5).
Los flavonoides son antioxidantes importantes con diversos efectos sobre la salud humana, entre ellos, efectos antivirales, anticancerígenos, antiinflamatorios y antialérgicos (6), que han sido estudiados para tratar enfermedades como el Alzheimer, la aterosclerosis, el cáncer, entre otros (7). Otras investigaciones se han centrado en entender el mecanismo de síntesis de estos compuestos, y así buscar vías para su sobreproducción en sistemas vegetales. Dentro de su ruta biosintética, la enzima PAL juega un rol clave, debido a que es la primera enzima de la ruta de los fenilpropanoides que eventualmente genera flavonoides (8). En fresa, se reportan 6 secuencias de los genes PAL, denominados FaPAL1, FaPAL2, FaPAL3, FaPAL4, FaPAL5 y FaPAL6 (9).
En trabajos previos realizados en el Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional (CINVESTAV-IPN) de Irapuato, se ha investigado la respuesta de los genes FaPAL2 y FaPAL6 de F. x ananassa cv. ''Camino Real”, y se encontró que estos eran expresados específicamente por iluminación con luz UV-C (10), lo que propone una vía para la sobreexpresión del gen PAL y producción de flavonoides. Sin embargo, se desconoce la funcionabilidad de las regiones río arriba del gen FaPAL2, principalmente porque no se reportan las secuencias.
Se han propuesto varias metodologías para el aislamiento de promotores genéticos, siendo una de ellas el PCR térmico asimétrico entrelazado, o TAIL PCR, desarrollado por Liu y Whittier (11), donde la amplificación se lleva a cabo entrelazando ciclos de PCR de alta rigurosidad con ciclos de rigurosidad baja, empleando primers degenerados y específicos para amplificar las secuencias objetivo de manera preferencial. Esta técnica se ha empleado exitosamente para el aislamiento de regiones promotoras de tres genes PAL de la planta D. bulbifera (12).
El presente trabajo de investigación tiene como finalidad identificar regiones promotoras del gen FaPAL2 de Fragaria x ananassa cv. ''Camino Real'' con el fin de evaluar su funcionabilidad en respuesta a la luz UV-C en frutos de fresa.
Metodología
Todos los experimentos fueron realizados en el Laboratorio de Bioquímica y Biología Molecular de Productos Naturales de Plantas del CINVESTAV-IPN Irapuato, durante el segundo semestre del 2019.
Material vegetal
Los frutos de fresa de la variedad se colectaron del invernadero del CINVESTAV-IPN, intentando cosechar aquellos que estuvieran iniciando el proceso de maduración. Se cosecharon frutos rojos, blancos y de coloración intermedia realizando un corte de 5 cm del pedúnculo, y para la agroinfiltración, se lavaron con agua desionizada estéril, después con una solución de cloro al 5% y, una vez en cámara de flujo laminar, un lavado con etanol al 70%, y último enjuague con agua desionizada estéril.
TAIL PCR
Se extrajo el material genético de hojas de F. x ananassa cv. ''Camino Real'' empleando el kit de E.Z.N.A. Plant DNA DS Mini Kit (Omega Bio-Tek, EUA), aplicando la elución en buffer TE. Con el material extraído se realizó un TAIL PCR siguiendo la metodología de Terauchi y Kahl (2000), empleando los 5 primers aleatorios ''forward'' propuestos por los autores, y 3 primers específicos ''reverse'' diseñados en el programa Primer3Plus para una región del gen FaPAL2 cerca del inicio de la transcripción (cuadro 1).
Para el primer ciclo de PCR se utilizó como templado ADN genómico extraído de hoja de fresa, en el segundo ciclo se utilizó como templado una dilución 1/10 del producto del primer ciclo de PCR, y en el tercer ciclo se empleó el producto del segundo ciclo de PCR sin diluir. Para cada ciclo se trabajaron 5 muestras distintas, una por cada primer forward, además, en el primer ciclo de PCR se empleó el primer reverse A, en el segundo ciclo el primer B, y en el tercer ciclo el primer C.

Transformación de Escherichia coli TOP10 con el vector pCR4-TOPO modificado
Los productos del tercer ciclo de PCR obtenidos en la metodología anterior se analizaron mediante una electroforesis para determinar las muestras con mayor cantidad de fragmentos separados. Estos se clonaron en un vector pCR4-TOPO, empleando el Kit de Clonación de TOPO TA para secuenciación. Siguiendo el protocolo suministrado por el Kit, se prepararon reacciones de 6 μL para cada uno de los productos del TAIL PCR.
Los plásmidos TOPO® con los fragmentos de interés se utilizaron para la transformación de células de E. coli quimiocompetentes. Para esto, se añadieron 10 μL de plásmido a un tubo con 100 μL de células competentes. Los tubos se mantuvieron en baño de hielo durante 60 minutos, seguidamente se colocaron en termobloque a 42°C durante 3 minutos, y se regresaron al hielo durante 10 minutos. Transcurrido este lapso, se añadió a cada tubo 300 μL de medio LB sin antibióticos, y se incubaron a 37°C durante 60 minutos, a 140 rpm. Finalmente, se tomaron alícuotas de 200, 100 y 50 μL de cada tubo, y se sembraron en placas Petri con medio LB semisólido suplementado con kanamicina (100 mg/mL) y estreptomicina (200 mg/mL), y se incubaron a 37°C durante toda la noche. La confirmación de transformantes se realizó mediante un PCR de colonia, esperando obtener fragmentos similares a los obtenidos en el tercer ciclo del TAIL PCR. C
Purificación de los fragmentos del TAIL PCR del vector pCR4-TOPO
Se realizó una extracción de plásmido de los cultivos transformados con el fragmento de interés, utilizando el Kit PureLink™ Quick Plasmid Miniprep de Invitrogen™. Una vez obtenidos los plásmidos purificados, estos se enviaron a secuenciar a Macrogen Inc. (sucursal de Corea del Sur) y, por otro lado, se procedió a amplificar el constructo genético que será clonado en el vector pBI121, y debido a que se desconoce la posición en la que el fragmento se introduce en el vector TOPO, se diseñaron 2 pares de primers distintos (cuadro 2), para asegurar que el extremo que contenga el ATG de iniciación del fragmento de interés se inserte de modo que quede flanqueando el gen GUS del vector pBI121. De las 2 reacciones de PCR, solamente una deberá generar amplicones.
Cuadro 2 Secuencias de primers utilizados en la PCR para amplificar los fragmentos de interés de los plásmidos pCR4-TOPO.

Transformación de Agrobacterium tumefaciens LBA4404 con el vector pBI121 modificado
Tanto el constructo de interés aislado en el paso anterior, como el plásmido pBI121 fueron digeridos con las enzimas BamHI y HindIII en reacciones de 20 μL, y dichas reacciones se utilizaron en la ligación de ambas fracciones genéticas, en reacciones de 20 μL, siguiendo los protocolos del sistema de clonación de enzimas de restricción Anza™ de Invitrogen™. Este vector pBI121 modificado se empleó para transformar, en primera instancia, células de E. coli TOP10 quimiocompetentes, para multiplicar el vector, y después se realizó una Miniprep, para finalmente emplear los plásmidos en la transformación de A. tumefaciens LBA4404.
La solución de plásmidos se dializó en agua destilada estéril empleando membranas de diálisis de 0.025 μm de la marca MerckTM. Se recuperaron 7 μL del plásmido y se añadieron a un tubo con 40 μL de células electrocompetentes de A. tumefaciens; la mezcla se transfirió a una celda de electroporación, y esta se electroporó a 2.5 V. Se añadieron 450 μL de medio YEB, y se transfirió la mezcla a tubos de 1.5 mL, donde permanecieron en incubación a 28 °C a 140 rpm durante 3 horas. Finalmente se sembraron las células en placas Petri con medio YEB suplementado con kanamicina (50 mg/mL) y rifampicina (50 mg/mL), y se incubaron a 28°C durante 2 días. La confirmación de transformantes se realizó mediante un PCR de colonia.
Agroinfiltración de frutos de Fragaria ananassa cv. ''Camino Real”
Se emplearon las colonias transformantes de A. tumefaciens con el constructo genético de interés para preparar un cultivo líquido en medio YEB suplementado con kanamicina y rifampicina, hasta que este alcanzó una OD600 entre 0.8 y 1. Los cultivos se centrifugaron y el pellet se resuspendió en medio MMA hasta alcanzar una OD600 entre 2 y 2.4. El cultivo se recuperó empleando una jeringa de 1 mL estéril, y se introdujo verticalmente en la parte superior de los frutos cerca del pedúnculo, y se procedió a inyectar entre 0.4 y 0.8 mL de cultivo bacteriano. Los pedúnculos se sellaron con un algodón humedecido con agua desionizada estéril, los frutos se colocaron en frascos de vidrio, y se incubaron a 27 °C con un fotoperiodo de 16 horas luz / 8 horas oscuridad durante 7 días.
Tratamiento con luz UV-C
A partir del tercer día de incubación, se seleccionaron los frutos y se cortaron rodajas de aproximadamente 2 mm de grosor en cámara de flujo laminar. Las rodajas se incubaron con una solución de cefotaxima (500 mg/L) por 10 minutos, se les secó el exceso de solución con un papel toalla, y cada grupo de rodajas se separó en dos. Solamente uno de ellos se colocó en rejillas metálicas dentro de una cámara con lámparas fluorescentes UV-C de 15 W (Tecno Lite G15T8), y permanecieron en irradiación durante 14 minutos.
Evaluación del sistema reportero GUS
De cada grupo de rodajas, cada mitad (irradiadas y no irradiadas) se colocó por separado en un tubo cónico de 15 mL, y a este se le añadió la solución X-Gluc hasta cubrir los tejidos. Los tubos se cubrieron con papel aluminio, y se incubaron a 37 °C durante 18 horas. Posteriormente, se decantó el X-Gluc, se secó el exceso de solución, y se evaluó la presencia o ausencia de coloración azul en los tejidos.
Resultados
TAIL PCR
A partir de los ciclos de amplificación del TAIL PCR se obtuvieron los fragmentos amplificados como resultado del tercer ciclo que se observan en la figura 1.
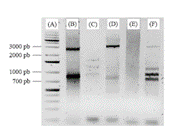
Figura 1 Electroforesis en gel de agarosa de la tercera reacción del TAIL-PCR. Corrida del marcador molecular GeneRulerTM 1 kb (A) y de los amplicones obtenidos con el primer reverse C y el primer forward AP103 (B), AP122 (C), AP138 (D), AP140 (E) y AP158 (F)
Purificación de los fragmentos del TAIL PCR del vector pCR4-TOPO recombinante
Se empleó el producto AP158 del TAIL PCR (figura 1.F) debido al menor tamaño de los amplicones, para facilitar su clonación en el vector pCR4-TOPO y transformación de E. coli. A partir de un PCR de colonia se confirmó que una colonia clonó el fragmento de interés de aproximadamente 1000 pb; se realizó la miniprep de esta colonia y PCR del plásmido con los primers M15Fw-C15Rv y C15Fw-M15Rv para definir la direccionalidad del inserto.
Se determinó que los primers M15Fw y C15Rv generaron el amplicón de interés, por lo que se purificó dicha banda y se analizó mediante una electroforesis (figura 2). Dicho fragmento se clonó en el plásmido pBI121 y el plásmido recombinante se empleó para transformar A. tumefaciens.
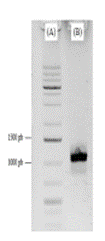
Figura 2 Electroforesis en gel de agarosa del fragmento de interés. Corrida del marcador molecular GeneRulerTM 1 kb (A), y el amplicón del vector AP158.J resultante de la purificación de la banda.
Evaluación del sistema reportero GUS en frutos de fresa agroinfiltrados
Se preparó el medio de agroinfiltración a partir de la colonia AP158.J de A. tumefaciens que contenía el constructo genético de interés (banda purificada del gel de la figura 2). La tinción histológica de los tejidos con X-Gluc después de 4 a 7 días de incubación se observa en la figura 3.
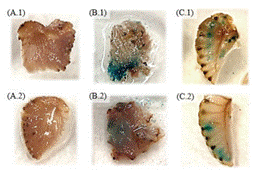
Figura 3 Expresión transitoria del gen GUS en frutos de fresa infectados con A. tumefaciens. Controles: Negativo sin irradiar (A.1), negativo irradiado (A.2), positivo sin irradiar (B.1), positivo irradiado (B.2). Agroinfiltrados: Con el constructo AP158.J sin irradiar (C.1), con el constructo P.AP158.J irradiado (C.2). La evaluación de los controles se efectuó en el día 4 y la de los agroinfiltrados en el día 7, posteriores al tratamiento de agroinfiltración.
Análisis in silico del promotor de FaPAL2
Con el resultado de la secuenciación del vector pCR4TM-TOPOTM ligado al fragmento AP158.J, se realizó una comparación in silico entre la secuencia obtenida con la secuencia del gen FaPAL2 (accesión KX450227.1) utilizando la base de datos de NCBI. Se determinó una coincidencia del 100% para la región situada corriente abajo del sitio de inicio de la transcripción, lo que comprueba que se aisló la región promotora del gen FaPAL2, generando una secuencia de 551 pb.
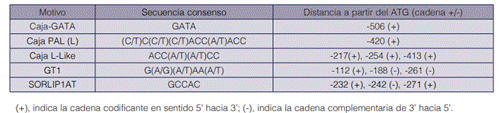
Esta secuencia se sometió a un análisis bioinformático para identificar elementos regulatorios relacionados con la respuesta a luz utilizando la plataforma y base de datos PLACE (https:// www.hsls.pitt.edu/obrc/index.php?page=URL1100876009). Como resultado de este análisis, se lograron identificar ciertos elementos, que se resumen en el cuadro 3.
Discusión
TAIL PCR
El ADN molde empleado en la primera reacción del TAIL PCR demostró tener la integridad y calidad idónea para el procedimiento ya que, como resultado de la tercera reacción del TAIL PCR, se obtuvieron múltiples amplicones, esto utilizando 4 de los 5 oligonucleótidos aleatorios. En la figura 1 se observan bandas definidas y sin barridos, que van desde los 700 a 3000 pb, sin embargo, se determinó que los primers AP103, AP138 y AP158 (figuras 1.B, 1.D y 1.F) generaron los amplificados deseables para su posterior clonación en un vector, ya que se consideran como buenos candidatos del promotor del gen FaPAL2. Esto basándose en otra investigación (13), donde compararon dos técnicas, entre ellas el TAIL PCR, para aislar la región promotora del gen RbcS de una microalga, esto porque el TAIL es uno de los métodos más utilizados para aislar regiones flanqueantes de secuencias conocidas, a pesar de que no se garantiza la ausencia de problemas por inespecificidad u obtención de bajas cantidades de amplicones.
Esta última afirmación puede explicar por qué el primer AP122 (figura 1.C) generó bandas sumamente tenues, y que el primer AP140 (figura 1.E) no generó amplificados. Importante destacar que la disminución en las diluciones de los productos de PCR para la segunda y tercera reacción evitó la generación de patrones manchados o amplicones tenues o barridos en la corrida de la tercera reacción, como los que se obtuvieron por (12) al correr en electroforesis los productos de la segunda reacción. Debido a que el ciclaje del TAIL se efectúa tanto en la segunda como en la tercera reacción para eliminar ruido de fondo y productos inespecíficos, esto sugiere que se requirieron productos de PCR más concentrados para eliminar dichos problemas en la tercera reacción.
Purificación de los fragmentos del TAIL PCR del vector pCR4-TOPO recombinante
Una vez obtenido y confirmado el amplicón de interés clonado en el vector pCR4-TOPO, fue necesario aplicar una estrategia de 2 pares de primers para amplificar los fragmentos y clonarlos con la direccionalidad correcta en el vector pBI121. El inserto está flanqueados por la secuencia del primer C (cuadro 1) en un extremo, y el primer AP158 en el otro; debido a que la secuencia de los insertos es desconocida, es imposible determinar en qué dirección se insertó el fragmento. Por otro lado, para asegurar que el ATG del fragmento se insertara en dirección al gen GUS del vector pBI121, se utilizó la secuencia de reconocimiento de las enzimas BamHI (flanquea posición de ATG) y HindIII (flanquea posición del promotor de FaPAL2).
De esta forma, si el fragmento se insertó en dirección AP-Inserto-primer C, debería de generar amplificados con los primers M15Fw y C15Rv (cuadro 2), contrario a si el fragmento se insertó en dirección primer C-Inserto-primer AP, caso en el que se amplificaría con los primers C15Fw y M15Rv (cuadro 2). El primer C15 empleado en ambos casos está flanqueado por la secuencia de reconocimiento de BamHI, de forma que, independientemente de la dirección del fragmento, al digerirlo con esta enzima, se asegura que el sitio de inicio de la transcripción (ATG) se inserte de modo que quede corriente arriba el gen GUS en el plásmido pBI121. Esto se ejemplifica de manera más gráfica en la figura 4.
Al obtener una amplificación positiva con los primers M15Fw y C15Rv al amplificar el vector AP158.J, se confirma que el fragmento se insertó en dirección primer AP-Inserto-primer C. Este fragmento, de aproximadamente 1000 pb, coincidió con los resultados previos de TAIL PCR y PCR de colonia, por lo que se descarta la generación de productos inespecíficos durante la PCR y, asociado a esto, se comprueba que la concentración de los primers y de la enzima utilizada, resultó ser la óptima para la amplificación, tal y como afirma en Díaz et al. (14).
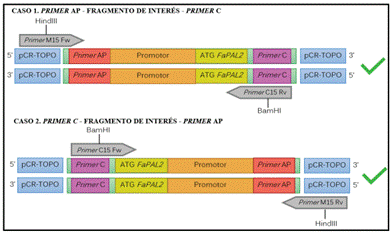
Figura 4 Representación gráfica de los casos de amplificación para los fragmentos de interés dentro del vector pCR4TM-TOPOTM.
Este resultado permitió determinar que la amplificación generada consiste en el constructo de interés con los elementos genéticos necesarios para ser introducido en el vector pBI121, por lo que fue necesario purificar dicha amplificación, ya que el producto de PCR no puede utilizarse directamente en la digestión o ligación porque presenta otros fragmentos y secuencias. Al cortar y purificar la banda, se obtiene un producto limpio de PCR, y al analizarlo mediante electroforesis, se obtiene una banda que garantiza la integridad de únicamente la construcción genética de interés, la cual se observa en la figura 2.
Evaluación del sistema reportero GUS en frutos de fresa agroinfiltrados
Los ensayos de expresión transitoria del gen GUS generaron resultados exitosos, a pesar de la variabilidad en el tamaño y madurez de los frutos de fresa. Como se observa en la figura 3.A, los controles negativos no generaron un cambio de coloración en la solución de X-Gluc, lo que indica la ausencia del gen GUS. Contrariamente al viraje de color que se obtuvo en el control positivo (figura 3.B), lo que manifiesta la expresión de la β-glucuronidasa codificada por el gen GUS, que hidrolizó el 5-bromo-4-cloro-3-indolil-β-D-glucorónido o X-Gluc (15, 16). Estos resultados definen la confiabilidad del método para determinar la expresión del gen de interés, en este caso, promotores que responden a luz UV. Este sistema de gen reportero ha sido ampliamente utilizado en plantas transgénicas, permitiendo determinar de la expresión genética espacial y temporal (17).
Con respecto a los frutos transgénicos, transformados con la construcción AP158.J, en todos, a excepción de uno, se detectó una expresión positiva de GUS (figura 3.C). La expresión negativa se atribuye a una limitante persistente a lo largo del protocolo relacionada con la integridad de los frutos, lo cual pudo repercutir en la ausencia de coloración azul. Esta dificultad suele ocurrir en la transformación de frutos carnosos, debido a la presencia de grandes células parenquimáticas, lo cual puede fácilmente alterar los tejidos al manipularlos (18), con jeringas, por ejemplo.
Por esta razón, el ablandamiento de los frutos presentó un problema durante la tinción histológica, esto se presentó especialmente en el fruto que se evaluó al día 5 tras la agro-infiltración, ya que, al momento de la cosecha, este se encontraba más rojo y de menor tamaño, lo que conllevó a una mayor alteración fisiológica al momento de la infección. Resultados distintos se presentaron con los dos frutos evaluados al día 5 tras la agro-infiltración; en la figura 3.C se denotan los spots azules de GUS, en una muestra que provenía de un fruto que, al momento de cosechar, tenía una coloración entre blanca-roja, y eran de mayor tamaño. Estos resultados coinciden con las afirmaciones de Zhao et al. (19), quienes explican que la optimización de la expresión transitoria de genes de interés en frutos en fresa está condicionada a factores como la temperatura, periodo de incubación, y estado de desarrollo del fruto, este último factor influyó en gran medida en los resultados obtenidos.
Por otro lado, al comparar los tejidos transformados sin y con exposición a la luz UV, no se reportó diferencia aparente en la actividad de GUS entre ambos frutos, esto se puede notar con la presencia de puntos azules tanto en frutos irradiados y como en no irradiados, específicamente en las muestras de las figuras 3.C.1 y 3.C.2. Sin embargo, la detección de este sistema reportero no puede compararse de forma precisa, ya que es meramente una evaluación colorimétrica visual, por lo que la actividad de GUS no puede medirse con precisión. Se han obtenido resultados similares con la detección de β-glucuronidasa al transformar frutos de fresa con la construcción pBI121-PrFaPAL6-GUS (20), y esto se justificó al citar que la expresión transitoria de frutos presenta limitaciones en cuanto a la observación de resultados, uno de ellos es el efecto variado entre frutos individuales que propicia una comparación limitada entre controles y tratamientos, la causa principal parece ser la variación en el grado de expresión de cada fruto de forma individual (19).
Es de destacar que, al no contar con diferencias notorias entre los tratamientos, no es posible atribuir la expresión del gen GUS a la presencia de promotores que responden de manera exclusiva ante luz UV. Por lo que es necesario analizar las secuencias clonadas en los plásmidos pCR4-TOPO recombinantes.
Análisis in silico y de la funcionalidad del promotor de FaPAL2
Como resultado del análisis in silico de la secuencia aislada del promotor FaPAL2 se identificaron varios elementos en cis asociados a la regulación de genes por exposición a luz, que se muestran en el cuadro 3. En el caso de la GATA-box y el motivo GT1, se ha comprobado que ambos están involucrados en la regulación por luz, y la combinación de ambos es necesaria para la respuesta a este estímulo (21). Sin embargo, ninguno de estos son elementos exclusivos para promotores de genes regulados por luz, ya que, por ejemplo, se encuentra también en el promotor CaMV 35S (22).
También se identificó la caja PAL (L); hay estudios que reportan que este es un sitio putativo del promotor del gen PAL-1, y que aparecen huellas de este después de tratamientos con luz (23), sin embargo, hay otros estudios que afirman que, si bien es un elemento necesario y exhibe expresión basal alta, no posee la actividad genética suficiente para ser inducido por exposición a la luz (24).
Por otro lado, también se identificó la Caja L-Like, que son distintas variaciones de la caja L, que también se han comprobado ser elementos regulatorios que responden a la luz (25). Y, por último, se identificó el motivo SORLIP1, que se ha identificado en promotores de genes inducidos por luz en arroz y Arabidopsis sp., y se ha lograron identificar este motivo en los promotores de los genes PAL1 y PAL2 provenientes de durazno (26, 27, 28).
La identificación de todos estos motivos permitió determinar que el promotor de FaPAL2 puede estarse activando mayormente por la exposición a luz, incluyendo UV-C, pero este estímulo no es un factor exclusivo que propicia su activación, lo cual apoya los resultados de la expresión transitoria de GUS discutidos en el punto anterior. Asociado a esto, se ha demostrado que la expresión de genes PAL responde a otros factores abióticos, por ejemplo, se ha estudiado el efecto sinérgico que tiene el daño mecánico con tratamientos de luz UV en la síntesis de compuestos fenólicos en zanahorias, y se determinó que la actividad de PAL aumentó en zanahorias cortadas y expuestas a luz UV-C, contrario a lo que ocurrió en zanahorias enteras (29). Por otro lado, se ha comprobado que la irradiación con luz UV-C de frutos de fresa promueve la expresión de FaPAL6, pero durante un periodo corto de tiempo, ya que no se detectó una expresión positiva después de 20 minutos de exposición al tratamiento (10).
Estos últimos estudios pueden sugerir que el daño mecánico efectuado al rebanar los frutos de fresa para su posterior tinción con X-Gluc pudo intervenir en el estudio de la inducción exclusiva del tratamiento con luz UV-C. Adicionalmente, el tiempo empleado para evaluar la expresión de GUS en frutos tratados y no tratados, pudo haber sido muy prolongado, lo que impidió observar diferencias de expresión entre ambos tratamientos.
Por otra parte, en relación con el promotor CaMV 35S, que es ampliamente utilizado en experimentos de transformación, se ha comprobado su alta actividad en órganos florales y polen de plantas transgénicas de fresa (30). Este promotor manifestó efectivamente una mayor actividad que el promotor de FaPAL2, lo que se evidenció comparando la tinción del control positivo y los frutos transgénicos (figura 3), ya que en los últimos las áreas teñidas se notan en menor proporción.
Conclusiones
La técnica TAIL PCR permitió aislar secuencias corriente arriba del gen FaPAL2, que se pudieron asociar a promotores y motivos que tienen una respuesta positiva a luz UV-C. Además, la técnica de electroporación probó ser más eficiente para la transformación de células electrocompetentes de A. tumefaciens, en comparación con la transformación química de células quimiocompetentes de E. coli.
El fragmento aislado corriente arriba del gen FaPAL2 demostró ser funcional como promotor, ya que promovió la expresión de β-glucuronidasa en frutos transgénicos de fresa. Sin embargo, al no observar diferencia de dicha expresión en los tratamientos empleados, se sugiere que el promotor está siendo activado de forma basal, y asociado a este resultado destacan el daño mecánico a los frutos y el tiempo prolongado de evaluación del sistema reportero GUS, que ambos limitaron la evaluación de la influencia exclusiva de la exposición a luz UV-C sobre el promotor de FaPAL2.