











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Las plantas poseen una gran variedad de productos naturales para defenderse de plagas y depredadores (1). Estos compuestos son productos del metabolismo secundario de la planta y poseen aplicaciones comerciales o de alto potencial terapéutico. Un ejemplo de estos compuestos son los terpenos, metabolitos secundarios muy diversos y que poseen diversas propiedades de interacción de la planta con el ambiente. De esta forma se relacionan con atractivos de polinizadores y repelentes de herbívoros (2). Aquellas plantas que acumulan productos naturales específicos se han utilizado durante cientos de años como medicamentos herbarios, por ejemplo, el ajenjo dulce (Artemisia annua) que se aplica para el tratamiento de la malaria (3). Debido al descubrimiento de nuevas dianas de fármacos, al surgimiento de nuevas enfermedades y a la creciente tolerancia microbiana a fármacos, la industria farmacéutica necesita constantemente nuevos metabolitos para combatir estas enfermedades (3). Uno de estos compuestos de importancia comercial es el farneseno (4). Este sesquiterpenoide está compuesto por tres unidades de isopreno y sus precursores y compuestos derivados se han relacionado con diversas actividades de aplicación médica, tal es el caso de la artemisina (4) (5)
Con el avance tecnológico de los últimos años, se han desarrollado nuevas alternativas, tanto para el tratamiento adecuado de los residuos, así como para su reutilización mediante la transformación de sus compuestos en productos de interés. Un ejemplo claro de este avance tecnológico es la biología sintética. Esta se define como la síntesis de moléculas de manera natural que se encuentran en pequeñas cantidades en la naturaleza o del todo no se producen de forma natural. La técnica se basa en el diseño de sistemas biológicos, a manera de reestructuración a novo (6). La biología sintética busca desarrollar nuevos organismos que pueda fungir como pequeñas fábricas capaces de procesar un compuesto (materia prima) y convertirlo en otro. Las principales herramientas de la biología sintética son: bioinformática, ingeniería metabólica y biología molecular (7).
Al aplicar principios de biología sintética al manejo de residuos, se obtienen una gran cantidad de aplicaciones potenciales. Una que resulta de gran interés es la generación de precursores de biocombustibles, vacunas, aceites y otros productos. Mediante el rediseño de rutas metabólicas se pueden generar microorganismos capaces de poder transformar cadenas largas de carbono como lo es la celulosa, componente estructural de gran abundancia en las plantas, en terpenos, tales como el farneseno. Este sesquiterpeno es precursor para la generación de biodiesel que puede usarse en automóviles, camiones de carga, buses o en generadores de energía. Mediante la implementación de diferentes genes las bacterias podrían ser capaces de degradar la celulosa en azúcares más simples y luego alimentarse de estos. La ruta metabólica sería direccionada a la producción de isopentil pirofosfato para luego producir directamente farneseno a través de la ruta del mevalonato (5).
Otro punto importante en la generación de terpenos de manera biosintética es la escogencia de un organismo que pueda expresar de forma idónea la ruta metabólica a insertar con el fin de poder generar de manera eficiente el compuesto deseado. Si bien Saccharomyces cerevisiae ha sido uno de los microorganismos por excelencia para la generación de farneseno, también se han generado diferentes investigaciones utilizando bacterias E. coli, la cual ha sido “ingenierizada” para poder producir alpha-farneseno insertando la ruta del mevalonato utilizando alpha farneseno sintasa proveniente de Malus doméstica, isopentil pirofosfato y dimetil alil pirofosfato, y farnesil difosfato sintasa (9). En investigaciones anteriores, Zhu y colaboradores, diseñaron una ruta metabólica sintética para generar farneseno (5).
En este estudio se diseñó una ruta metabólica in sílico la cual fue optimizada para la producción de Farneseno e insertada en E coli. Un total de 8 genes fueron ensamblados juntos a promotores para iniciar la transcripción, un sitio de unión con la ribosoma para iniciar la traducción a partir del transcrito y dos terminadores para detener la traducción.
Metodología
Diseño de genes sintéticos
Los genes fueron diseñados y optimizados mediante paquetes bioinformáticos tales como MetaCyc para la obtención de la ruta metabólica del farneseno (8). Posteriormente las secuencias de los genes involucrados fueron obtenidas a través de la base de datos del Centro Nacional de Información para Biotecnología (NCBI) de Estados Unidos. Estas secuencias fueron optimizadas para ser expresadas en Escherichia coli utilizando la interfase Optimizer (10). Posteriormente cada gen fue diseñado para tener un prefijo corriente arriba y un sufijo corriente abajo, las cuales estarían flanqueando el gen de interés y que permiten el ensamblaje entre sí y con otras piezas genéticas incluyendo: promotor, sitio de unión a ribosoma (RBS) y doble terminador para conformar un gen funcional. Los arreglos de cada secuencia se hicieron mediante el programa Gene Designer (11). Las secuencias codificantes (CDS) fueron sintetizados por la empresa GeneScript.
Ligación de genes para desarrollo de Bio-bloques activos
Para poder ligar los genes con promotores, un RBS y un doble terminador se utilizó el sistema de ensamblaje RFC10. Primero se unió cada uno de los genes con un doble Terminador y en paralelo un RBS con un promotor, una vez que cada una de estas piezas estaban ensambladas se acopló el Promotor+RBS con CDS+Doble terminador para tener un gen funcional. Promotores y CDSs fueron digeridas como un biobrick corriente arriba y RBS y Doble terminador como una piezas corriente abajo. Además se digirió enzimáticamente un plásmido de tal forma que pudiese recibir ambas piezas genéticas y que tuviese diferente gen de resistencia a los plásmidos que contenían las piezas mencionadas.
Para digerir cada una de las piezas se mezclaron 250 ng de plásmido de cada una de las piezas y 0,5 ul de cada una de las correspondientes enzimas; EcoRI y SpeI en el caso de las piezas corriente arriba y XbaI y PstI en las piezas corriente abajo. En el caso del plásmido destino se agregaron 125 ng de ADN. Además se agregaron 5 ul de CutSmart y se llevó a un volumen final de 20 ul con agua libre de nucleasas. Las reacciones se incubaron a 37 ºC por 3 horas. Al finalizar este lapso se colocó a 80 ºC por 30 minutos para inactivar la reacción.
Posteriormente las piezas fueron ensambladas entre sí de manera enzimática. Para esto se trabajó en una relación 4:4:1 pieza corriente arriba, pieza corriente abajo y plásmido. Para obtener las cantidades correctas de cada pieza se utilizó la herramienta NEBioCalculator v1.7.1.
Transformación bacteriana con los bio-bloques obtenidos en un solo constructo
Para ensamblar los diferentes constructos se utilizó un plásmido que contenía una proteína roja fluorescente (RFP) en el sitio de clonaje. Se transformó la cepa DH5α de E. coli con las piezas genéticas ensambladas. Para esto se adicionaron 5 μl de la ligación a un tubo con 50 μl de células competentes y se mezcló el contenido con la misma micropipeta. Las células fueron incubadas con la mezcla de transformación por 30 minutos. Luego se realizó un choque térmico transfiriéndolos primero a un baño maría e incubando a 42 °C durante 90 segundos y posteriormente en hielo por 2 minutos. Una vez realizado el choque térmico; se adicionaron 200 μl de medio SOC líquido precalentado a 37º C en cada tubo de células a transformar y estos se incubaron por 45 min a 37 °C/120 rpm. Transcurrida la incubación, se transfirieron 75 μl y se inocularon por extensión en placas distintas con medio LB suplementado con agar y el antibiótico correspondiente. Las placas fueron incubadas a 37° C por un lapso de 12-16 horas aproximadamente. Al día siguiente se tomaron las colonias que crecieron en el medio selectivo con antibiótico y se pasaron a un nuevo medio de cultivo en placa. De esta placa nueva se tomó 1 colonia bacteriana y se inoculó en 5 ml de medio LB con 50 ul de antibiótico en un tubo de 50 ml, esto para extraer los plásmidos que contienen las construcciones previamente hechas.
Para la extracción y purificación de plásmidos cada muestra fue incubada a 37º C/120 rpm por 12-16 horas. Transcurrido el tiempo de incubación las muestras fueron centrifugadas a 8000 rpm por 10 minutos. El pellet obtenido fue resuspendido en 250 μl de la solución de suspensión. A esto se le agregaron 50 μl de la solución de lisis y se mezcló hasta homogenizar. Posteriormente se agregaron 350 μl del buffer de neutralización, se mezcló por inversión para luego inmediatamente centrifugar por 10 min a 11000 rpm. Luego, se colocó el sobrenadante en columnas de filtración y se centrifugó la mezcla por 1 min a 11000 rpm y posteriormente se descartó el líquido filtrado. Seguidamente se agregaron 500 μl de la solución de lavado a la columna y se centrifugó nuevamente por 1 min a 11000 rpm. Finalmente, se centrifugó la columna por 2 min a 11000 rpm sin agregarle ningún reactivo. Se agregaron 30 μl de agua libre de nucleasas y se centrifugó la columna por 1 min a 11000 rpm. Al final, los minipreps obtenidos fueron almacenados -20 C.
En paralelo, este procedimiento se repitió para el ensamblaje de promotores y RBS (ribosomal binding site) siendo los promotores digeridos como piezas corriente arriba y el RBS como una pieza corriente abajo. El plásmido receptor utilizado presentaba resistencia a cloranfenicol, diferente a las piezas a ensamblar que estaban en plásmidos con resistencia a ampicilina. Para modular el sistema de expresión se utilizó el promotor inducible E-Omp R con código iGEM R0082. El RBS utilizado fue el B0034, descrito por Michael Elowitz (12).
Una vez que se obtuvieron plásmidos purificados de cada uno de los promotores ensamblados por separado con el RBS de Elowitz (promotor+RBS), se procedió a ensamblar promotores+RBS con genes más doble terminador (genes+DT). Para esto, la combinación de promotor+RBS fue digerida como una pieza corriente arriba mientras que las construcciones genes+DT fueron digeridas como piezas corriente abajo. Después de digeridas fueron ligadas entre sí y las mismas posteriormente ligadas a un plásmido previamente digerido con resistencia a ampicilina. Posteriormente la ligación fue utilizada para transformar cepas E. coli DH5α como se describió previamente. Las bacterias fueron finalmente sembradas en medio con ampicilina.
Posteriormente, cada bacteria transformada fue cultivada como se describió previamente para obtener su respectivo plásmido. Una vez obtenido el plásmido se le realizó una prueba de PCR con dos juegos de imprimadores diferentes para confirmar que el gen completo estuviese presente.
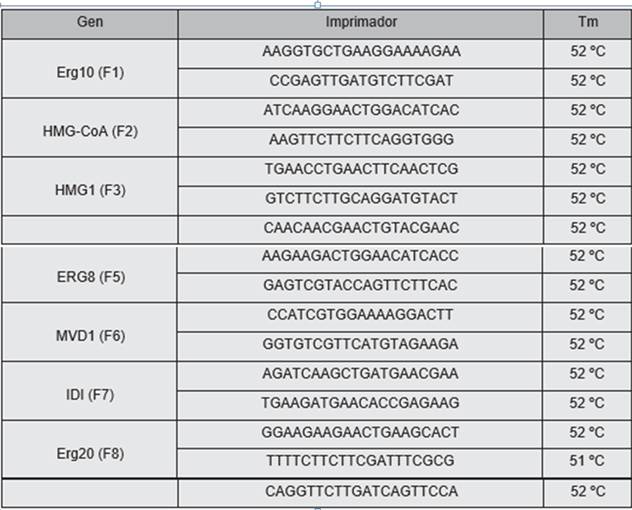
Para la confirmación de los ensamblajes realizados los genes fueron amplificados mediante PCR utilizando dos juegos de imprimadores (cuadro 1). V2F y VR que amplificaron desde el plásmido y HprF y HprR que flanqueaban el constructo ya que fueron diseñados a partir del prefijo y sufijo. Los imprimadores utilizados fueron los que se anotan en el cuadro 1.
Cuadro 1 Imprimadores diseñados para la caracterización de genes sintéticos
| Imprimador | Secuencia | Tamaño |
| HprF | Gaattcgcggccgcttctag | 15.8 kb aprox |
| HprR | Tactagtagcggccgctgcag | |
| VF2 | Tgccacctgacgtctaagaa | 16.0 Kb aprox |
| VR | attaccgcctttgagtgagc |
Luego de verificar por PCR los genes completos, se procedió a ensamblar los genes entre sí para poder armar la ruta metabólica completa. La digestión de las diferentes piezas ser realizó según se describe en el cuadro 2.
Cuadro 2 Digestión de genes completos para el ensamblaje de la ruta del mevalonato.
| Gen | Tipo de digestión |
| Erg10 (F1) | Corriente arriba |
| HMG-CoA (F2) | Corriente abajo |
| HMG1 (F3) | Corriente arriba |
| ERG12 (F4) | Corriente abajo |
| ERG8 (F5) | Corriente arriba |
| MVD1 (F6) | Corriente abajo |
| IDI (F7) | Corriente arriba |
| Erg20 (F8) | Corriente abajo |
Medición de Expresión de los genes mediante PCR Tiempo Real
Para corroborar el ensamblaje de la ruta biosintética del mevalonato se midió la expresión de cada uno de los genes mediante PCR en Tiempo Real. Para esto se extrajo ARN y se transformó en ADN copia, mediante retrotranscriptasa inversa. Primero se cultivó E. coli en medio LB suplementado con ampicilina y se incubó a 37 ºC durante 12 horas.
Para extraer ARN se utilizó el kit SV Total RNA Isolation System de la marca Promega. El ARN obtenido se almacenó a -80 ºC.
Para convertir el ARN en ADN copia se siguieron las instrucciones especificadas por el kit utilizado (GoScript Reverse Transcription System).
Para la realización del PCR Tiempo Real se preparó la mezcla de PCR siguiendo protocolos estandarizados y el uso de la master mix provisto por la casa fabricante. Para esta mezcla se utilizó un par de cebadores para cada uno de los genes diseñados según el cuadro 3.
Para la amplificación de las muestras se utilizó la sonda Syber Green. La temperatura específica de anillamiento para cada par de cebadores se encuentra en el cuadro 5.
Resultados
Diseño de genes sintéticos mediante paquetes bioinformáticos
Se obtuvieron 8 genes sintéticos ensamblados en un plásmido pUC19 para su posterior transformación en E. coli DH5α
Ensamblaje de genes para generación de Bio-bloques activos.
Los genes diseñados y sintetizados fueron todos ensamblados con diferentes promotores con el fin de poder evaluar la posible respuesta de E. coli ante la expresión de proteínas recombinantes y así mejorar los rendimientos de producción de farneseno. Los tamaños de los diferentes genes dependiendo del promotor utilizado se pueden observar en el cuadro 4.
Cuadro 3 Genes obtenidos mediante diseño bioinformático, sintetizados a través de Genescript.
| Gen | Función | Origen |
| Erg10 (F1) | Acetoacetil-CoA sintetasa | S. cerevisiae |
| HMG-CoA (F2) | Hidroximetilglutaril-CoA sintasa | Malus sp. |
| HMG1 (F3) | HMG-CoA reductasa | Malus sp. |
| ERG12 (F4) | Mevalonato kinasa | S. cerevisiae |
| ERG8 (F5) | Fosfomevalonato kinasa | S. cerevisiae |
| MVD1 (F6) | Isopentenil difosfato | S. cerevisiae |
| IDI (F7) | Isopentenil difosfato | Malus sp. |
| Erg20 (F8) | E,E-alpha-farneseno | Malus sp. |
Cuadro 4 Tamaño esperado de genes construidos en Gene Designer
| Gen | Tamaño (pb) | Gen | Tamaño (pb) | Gen | Tamaño (pb) |
| AF1 | 1525 | AF3 | 3497 | AF6 | 1523 |
| CF1 | 1434 | EF3 | 3455 | BF6 | 1573 |
| EF1 | 1487 | AF4 | 1528 | BF7 | 1249 |
| AF2 | 1808 | BF4 | 1578 | AF8 | 3143 |
| CF2 | 1713 | BF5 | 1738 | BF8 | 3193 |
| EF2 | 1766 | DF5 | 1593 |
Transformación bacteriana con los bio-bloques obtenidos en un solo constructo
El uso de la proteína RFP como indicador de transformación, permitió que posterior a la digestión del plásmido para introducir el nuevo constructo, este remplazará a la RFP, así cuando los plásmidos se recircularon sin tener el constructo insertado le dieron una tonalidad roja a la bacteria. Cuando el plásmido y el constructo se ligaron de forma correcta este no dio coloración roja.
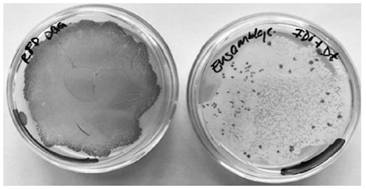
Figura 1 Transformación de E. coli DH5α. Izquierda, control positivo de transformación. Derecha, Ligación gen y doble terminador.
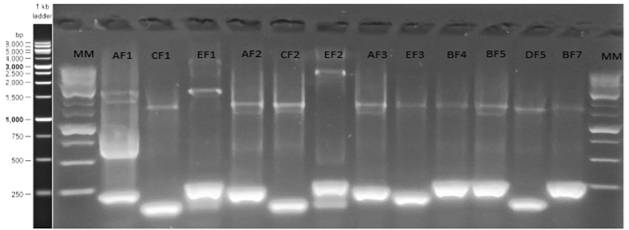
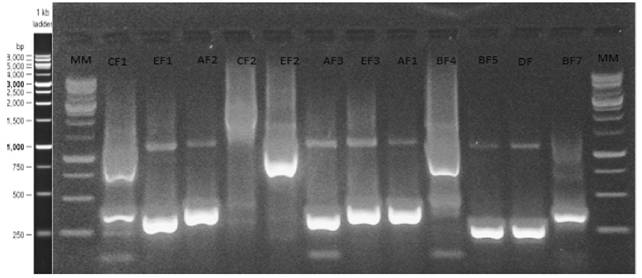
Según los resultados obtenidos con los PCR realizados, se verificó la presencia de los genes ensamblados, ya que los tamaños de las bandas de los amplicones coinciden con el tamaño esperado para cada uno y según los imprimadores utilizados (figuras 2 y 3 ).
Medición de la expresión de los genes mediante PCR Tiempo Real para la verificación de los componentes de la ruta metabólica del mevalonato.
Se realizó un análisis de la expresión de genes mediante el estudio del ARN, como una forma alternativa de verificar que la bacteria es potencialmente capaz de producir farneseno con la maquinaria sintética que se le insertó.
Para esto en el cuadro 5 se muestran los imprimadores utilizados para la reacción de PCR tiempo real.
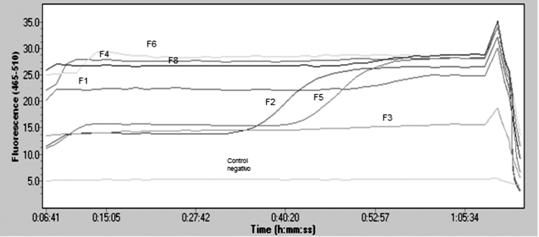
La figura 4 muestra el electroferograma observado en la reacción de RT-PCR (sonda SYBR- Green) para la expresión de los 8 genes contemplados en la ruta modificada del mevalonato que conduce a la expresión del farneseno.
Cuadro 5 Imprimadores específicos para la amplificación de genes componentes de la ruta biosintética del Mevalonato (MVA) por la técnica de RT-PCR
Discusión
Diseño y síntesis de genes y ensamblaje en biobloques.
La ruta metabólica diseñada y generada de manera sintética empieza con el gen ERG10 el cual traduce la enzima acetoacetil-CoA sintetasa que tiene una actividad enzimática de ligasa. Esta enzima se encarga de la conversión de cuerpos cetónicos en acetoacetil-CoA el cual se requiere para la síntesis de colesterol. Posteriormente se diseñó el gen ERG13 que genera la enzima hidroximetilglutaril-CoA sintasa (HMG-CoA sintasa). Esta enzima cataliza la reacción en la que el acetil-CoA se condensa con el acetoacetil-CoA para formar 3-hidroxi-3-metilglutaril- CoA (HMG-CoA). Seguidamente se diseñó y sintetizó el gen HMG1 que produce la enzima HMG-CoA reductasa. Esta enzima es dependiente de NADH/NADPH que controla la velocidad de la vía del mevalonato. Dentro de la ruta del mevalonato existe la subruta: biosíntesis del isopentil difosfato. Esta ruta está formada por los genes ERG12, ERG8 y MVD1. El gen ERG12 expresa la enzima mevalonato kinasa la cual contribuye a la regulación en las rutas metabólicas de isoprenoides y esteroles.
La mevalonato kinasa cataliza la conversión del intermediario mevalonato en (R)-5-fosfomevalonato. El gen ERG8 produce la enzima fosfomevalonato kinasa la cual es una enzima esencial del citosol celular. Esta enzima cataliza la conversión del (R)-5- fosfomevalonato (producto del gen ERG12) en (R)-5-difosfomevalonato. En ambas reacciones ejecutadas por los genes ERG12 y ERG8 media ATP, el cual cede un grupo fosfato en ambos productos. El gen MVD1 traduce para la enzima mevalonato pirofosfato decarboxilasa la actual actúa como un heterodimero y cataliza la conversión de (R)-5-difosfomevalonato en isopentenil difosfato. E. coli tiene una copia de IDI, el cual genera la enzima isopentil difosfato isomerasa, sin embargo, se inserta como parte de esta ruta sintética debido a que la sobreexpresión de este gen conlleva a una estrecha relación entre el consumo y la producción de IPP y DMAPP lo cual contribuye significativamente al aumento en la producción de α-Farnseno además de aliviar la inhibición en el crecimiento de E. coli.
Esta enzima cataliza el arreglo 1,3 alilico del sustrato (isopentil pirofosfato) en DMAPP que es su isómero más alilico. Como último gen se diseñó una fusión de dos genes: ERG20 y αFS, ya que la fusión de ambas proteínas ayuda a incremetar la concentración farnesil pirofosfato disponible de forma inmediata a α-farnesil sintasa y esta unión previene la difusión del farnesil pirofosfato en el citoplasma celular. El gen ERG20 traduce la enzima farnesil pirofosfato sintasa la cual cataliza la condensación secuencial de isopentenil pirofosfato con pirofosfato alilico, dimetilalil pirofosfato y luego con geranilpirofosfato hasta producir farnesil pirofosfato. Este último subproducto es posteriormente transformado por la proteína αfarnesil sintasa en (E,E)- αfarneseno. Una sobreexpresión de esta fusión de proteínas lleva al aumento de α-farneseno, sin embargo, lleva a la reducción de la biomasa. Este efecto no se da por una sobreexpresión de estas últimas proteínas o la mutación generada sino al desgaste de compuestos esenciales como DMAPP, Isopentil pirofosfato y farnesil pirofosfato. Cabe señalar, como se hizo previamente, la necesidad de sobreexpesar el gen IDI para aumentar la concentración de los compuestos esenciales ya mencionados y que haya suficientes precursores para generar α-farneseno sin comprometer el crecimiento bacteriano (9).
Como producto adicional se obtuvieron diferentes versiones de expresión de los genes ya que en lugar de un único promotor se utilizó el promotor ompR de regulación genética positiva. Este promotor se toma de la región corriente arriba de ompC. El OmpR fosforilado se une a los tres sitios del operador y activa la transcripción.
Transformación bacteriana con los bio-bloques obtenidos en un solo constructo.
Como se puede observar en la figura 1, hay colonias rojas y colonias blancas. Las colonias rojas corresponden a bacterias que fueron transformadas con plásmidos re-circularizados y de ahí su coloración por lo que estas colonias se descartan. Las colonias de color blanco corresponden a las bacterias que fueron tranformadas con el plásmido que contiene el inserto deseado. El constructo desplazó al gen fluorescente y de ahí que este perdiera su coloración. De esta forma las colonias blancas son pasadas a nuevas placas y las colonias rojas son desechadas ya que esta coloración indica que no fueron transformadas.
En las figuras 3 y 4 se podrá corroborar que los genes CF1, AF2, CF2, EF2, BF4, BF5, DF5 y BF7 poseen los tamaños esperados para ambos juegos de imprimadores. Contrario a esto los genes AF1, EF1 y BF7 presentan un tamaño correspondiente para los imprimadores HpR pero en el caso de los imprimadores V2F y VR no se obtuvieron amplificaciones. El gen AF3 presenta amplificación positiva para los marcadores V2F/VR.
En el caso de los genes que lograron amplificar por PCR con ambos imprimadores presentan una alta probabilidad de que correspondan a los genes deseados. En el caso de los genes que amplificaron con un solo juego de imprimadores, se puede tener una alta certeza de que estos genes corresponden a los buscados. Cabe resaltar que los imprimadores HPR están basados en los prefijos y sufijos de los genes mientras que los V2F/VR están basados en la secuencia del plásmido por lo que estos últimos pudieron sufrir alguna mutación en el proceso de digestión que afecte la amplificación por PCR.
Medición de la expresión de los genes de la ruta del farneseno mediante PCR Tiempo Real
El adecuado crecimiento de cepas de E. coli transformadas demostró que la maquinaria sintética insertada no afecta el correcto funcionamiento de la bacteria. Por otro lado, su posterior análisis mediante PCR Tiempo Real (Fig. 5) permitió demostrar el correcto ensamblaje de los genes en la bacteria y su adecuada expresión. El estudio comparativo de la expresión de los genes de la ruta biosintética del mevalonato mostró un incremento en los niveles de ARNm en todos los genes recombinantes insertados en E. Coli, sin embargo, el nivel de expresión varía entre los genes. Esto puede deberse a la complejidad de varios genes, sobre todo de ERG20 y αFS debido a la fusión entre ambos genes (13).
Conclusiones
- Se logró ensamblar la ruta metabólica que incluya todos los genes modulados por diferentes promotores.
- El sistema de rfc10 permite el ensamblaje de piezas de una forma eficiente, logrando tener genes completos que posteriormente pueden ensamblarse entre sí para generar rutas metabólicas completas.
- Se obtuvieron las secuencias de las enzimas que reportaban mejores rendimientos y se optimizaron para poder ensamblarlas a manera de biobloques. para ser expresadas en E. coli.
- Los genes sintéticos completos ensamblados con un doble terminador, un RBS y un promotor se expresaron correctamente en las bacterias transformadas.

