










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Ciencias Médicas
Print version ISSN 0253-2948
Rev. costarric. cienc. méd vol.26 n.1-2 San José Jan. 2005
Diagnóstico y tamizaje del retardo mental hereditario más común, situación en Costa Rica.
Isabel Castro-Volio1*, Rebeca Vindas-Smith1, Patricia Cuenca-Berger1.
Resumen
El síndrome del cromosoma X frágil (FRAXA) es la segunda causa genética de retardo mental y la forma más frecuente de retardo mental hereditario. FRAXA es causante de discapacidades que van desde grados variables de problemas de aprendizaje hasta retardo mental. Con frecuencia se asocian retrasos severos en el lenguaje, problemas de conducta, comportamiento semejante al autista, testículos agrandados, orejas grandes o prominentes, hiperactividad, retraso en el desarrollo motor y deficiente integración sensorial. Se hace un resumen del conocimiento actual de esta patología y del trabajo de los autores. Se tocan temas como el producto génico, los métodos de diagnóstico, el cuadro clínico, la epidemiología, la prevención, el tratamiento, el tamizaje y la situación en Costa Rica.
Palabras clave: genética humana, mutaciones inestables, síndrome del cromosoma X frágil, tamizaje en cascada, retardo mental.
Abstract
Fragile X syndrome (FRAXA) is the most common type of hereditary mental retardation, and the second commonest with genetic origin. The range of affection in FRAXA includes from learning problems to mental retardation. The syndrome includes speech and language deficits, abnormal behaviours, including autistic features, macro orchidism, prominent ears, hyperactivity, sensorial integration and motor impairments. Actual data and the authors own work is reviewed. Topics approached are the gene product, diagnostic methodology, clinical picture, epidemiology, prevention, screening and the actual situation in Costa Rica regarding this pathology.
Key words: Human genetics, unstable mutations, fragile X syndrome, cascade screening, mental retardation.
Introducción
El síndrome del cromosoma X frágil, conocido también como FRAXA, es la segunda causa genética de retardo mental y la forma más frecuente de retardo mental hereditario, con una frecuencia estimada de 1: 4 000 varones y 1: 6 000 mujeres (1). FRAXA es una anomalía hereditaria ligada al cromosoma X, causante de discapacidades que van desde grados variables de problemas de aprendizaje hasta retardo mental. Con frecuencia se asocian retrasos severos en el lenguaje, problemas de conducta, comportamiento semejante al autista, testículos agrandados, orejas grandes o prominentes, hiperactividad, retraso en el desarrollo motor y deficiente integración sensorial (2). Este síndrome es responsable de la mayoría de los casos de trastornos hereditarios en el desarrollo psicomotor. Obedece a un mecanismo mutacional desconocido hasta 1991 y llamado de varias maneras: mutaciones inestables, amplificación de ADN y expansión de tripletas repetidas (3-7). El defecto molecular consiste en una cantidad aumentada de repeticiones de la tripleta CGG (citosina guanina guanina) en una región no codificante del gen FMR1, situada en la porción terminal del brazo largo del cromosoma X. En la población general el tracto de repeticiones CGG es polimórfico, varía desde unas cuantas repeticiones hasta más de mil, con una moda de 29 repeticiones en tandem. Cuando se sobrepasa un umbral de alrededor de 200 repeticiones, se produce la metilación anormal de una isla CpG, elemento regulador que controla la transcripción de los genes. La metilación impide la transcripción del gen FMR1 y por lo tanto no se produce la proteína que éste codifica. Esta proteína se conoce como FMRP. Este síndrome segrega como dominante ligado al X con penetrancia reducida y expresión variable, afecta a individuos de ambos sexos, los cuales, cuando acarrean la mutación X frágil, pueden presentar deficiencia mental (8).
El producto del gen FMR1:
La proteína FMRP, producto del gen FMR1, está ausente en los pacientes afectados por FRAXA. Pesa 69 kDa y se producen muchas isoformas debido al corte y empalme (splicing) alternativo. Se expresa básicamente en el cerebro, específicamente en las neuronas, en los testículos, en la placenta y en los linfocitos. FMRP se liga selectivamente a ARNs mensajeros formando así un complejo de ribonucleoproteína mensajera que se asocia con los polirribosomas. Este complejo ejerce una función fundamental en las neuronas al regular la traducción de otras proteínas. La FMRP se localiza en las sinapsis y su ausencia altera la plasticidad sináptica, la cual está relacionada con el aprendizaje y la memoria. Su función es participar en el transporte de ARNs mensajeros desde el núcleo hacia los polirribosomas que están principalmente localizados en las dendritas proximales de las neuronas, donde participa en la traducción de las proteínas. Su afinidad es específica para ARNs que se expresan en células nerviosas, incluyendo su propio ARN. La proteína tiene una región conocida como caja RGG que es la responsable de la unión con ciertos ARNs mensajeros que a su vez tienen un motivo llamado cuarteto G que permite esta unión. El retardo mental es el resultado de anormalidades en la traducción de proteínas que dependen de FMRP para el transporte de sus ARNs y para su traducción (9). FMRP actúa como una represora de la traducción y regula negativamente la traducción de varios ARN específicos de dendritas, algunos de los cuales codifican para proteínas del citoesqueleto y moléculas para la transducción de señales, importantes para la plasticidad sináptica. Por tanto, en ausencia de FMRP se observan niveles aumentados de proteínas relacionadas con funciones neuronales. La inhibición de la traducción a través de FMRP parece ocurrir por medio de un mecanismo recientemente descubierto llamado interferencia por ARN, el cual es un proceso natural para silenciar la expresión génica (10,11). La interferencia por ARN parece ser un antiquísimo mecanismo evolutivo para proteger a los organismos de los virus que contienen ARN como su material genético. Tiene dos componentes principales: una enzima llamada Dicer que degrada el ARN viral de doble hebra y un complejo enzimático llamado RISC (RNA Induced Silencing Complex) que busca y destruye el ARN viral que posee las mismas secuencias del ARN degradado por Dicer. Este mecanismo parece tener otras funciones igualmente importantes, entre ellas, la de regular a niveles muy finos la expresión genética celular normal. Se está estudiando intensamente el papel que juega FMRP en el mecanismo de interferencia por ARN, pues aún quedan muchas interrogantes por resolver (11,12).
El diagnóstico del cromosoma X frágil:
El diagnóstico se realizaba inicialmente gracias a la expresión citogenética de un sitio frágil en Xq27.3 (13,14). Esta prueba diagnóstica ya no se utiliza más pues su sensibilidad y especificidad son insuficientes (15). Actualmente existen diferentes sondas específicas para hacer diagnóstico directo de la mutación en los afectados mediante la hibridación de Southern. Con esta técnica es posible determinar amplificaciones de 80 o más repeticiones. Cuando los familiares de un individuo afectado demandan consejo genético, se hace necesario usar la reacción en cadena de la polimerasa (PCR), para diagnosticar los portadores de amplificaciones pequeñas, ya que la PCR permite conocer exactamente el tamaño de la región amplificada. Combinando los dos métodos es posible discriminar entre el alelo normal (con un máximo de ~ 54 repeticiones de la tripleta CGG), el alelo de los portadores asintomáticos (con ~ 55 hasta ~ 200 repeticiones de la tripleta CGG) y el alelo de los afectados por la mutación (con más de ~ 200 repeticiones de la tripleta CGG). Otra tecnología útil es la secuenciación por fluorescencia láser automática. Estos métodos de laboratorio son muy exactos pero muy caros, de manera que no se ofrecen a la población general (16,17).
El cuadro clínico:
No es posible hablar de diagnóstico clínico de FRAXA, sino solamente de sospecha clínica, que debe ser confirmada mediante los estudios moleculares.
Las alteraciones fenotípicas que produce esta amplificación del ADN son diversas, y se enmarcan dentro del síndrome de cromosoma X frágil o síndrome de Martin-Bell, en honor a J.P. Martin y a J. Bell, quienes delinearon el síndrome por primera vez en 1943. El cuadro clínico a menudo difiere según el género del paciente. Las características físicas pueden no ser evidentes en afectados jóvenes y suelen ir apareciendo conforme van creciendo. Antes de la pubertad los varones con el síndrome pueden mostrar orejas protuberantes, paladar ojival, puente nasal aplastado, macrocefalia, pliegues epicánticos, un único pliegue palmar, mala coordinación de sus movimientos, articulaciones muy flexibles e hipotonía. Después de la pubertad se pueden incluir otros rasgos como cara larga y angosta con mentón prominente, orejas grandes, macro-orquidismo y prolapso de la válvula mitral. Adicionalmente pueden mostrar laxitud del tejido conectivo, problemas visuales, escoliosis, tics motores, mayor talla durante la niñez y menor durante la adultez, piel aterciopelada, pie equino varo, pliegue en la planta del pie, frente ancha o abultada, manos grandes, hernias, pulgares con doble articulación, pie plano y pecho excavado. Entre las características físicas de las mujeres sobresalen la cara angosta y las orejas grandes (2).
El fenotipo neurocognitivo también es diferente entre los géneros. Debido a la inactivación al azar de un cromosoma X en las mujeres, solamente el 30-50% de ellas va a manifestar síntomas cognitivos, a pesar de tener la mutación. Las que son sintomáticas y la mayoría de los varones tienen predisposición a presentar desde la infancia un conjunto de problemas cognitivos, conductuales y emocionales, que en términos generales incluyen: retardo cognitivo con coeficientes intelectuales (CI) que disminuyen con la edad, trastornos en el lenguaje y la comunicación, desarrollo anormal y reducido de los comportamientos de adaptación, anomalías cognitivas particulares dentro de los dominios de función ejecutora y cognición visual-espacial, hiperactividad y problemas significativos con sobre estimulación y ansiedad.
Las madres de bebés afectados, casi siempre varones, pueden notar desde que tienen nueve meses, atrasos en el desarrollo con tonicidad y coordinación motora anormales. Sin embargo, el diagnóstico suele postergarse hasta que cumplen tres años, como resultado de retrasos manifiestos en el habla o anomalías conductuales. En la edad pre-escolar su tasa de desarrollo suele equivaler a un tercio o un medio de la esperada para los niños de edad similar y desarrollo típico. El lenguaje expresivo se ve más afectado que el receptivo y está más atrasado, junto con el funcionamiento cognitivo, que las funciones motoras y de adaptación. Las curvas de desarrollo cognitivo y adaptativo crecen más rápidamente hasta los cinco años de edad aproximadamente, más lentamente hasta alrededor de los diez años y luego se estancan, con disminución en los puntajes de CI en los años puberales. Desde la edad pre-escolar hasta la adolescencia, los chicos muestran discrepancias cada vez mayores entre el nivel de lenguaje y la edad cronológica. También desde temprana edad es evidente el comportamiento autista, probablemente asociado a un sistema nervioso mal modulado, con aumento de la excitabilidad y problemas con inhibición y habituación. Las niñas muestran un desarrollo muy variable, con anomalías cuantitativa y cualitativamente menos severas. La presencia de ansiedad social, timidez y conducta de evitación en la edad escolar y pre-escolar, parece ser un factor de riesgo para la aparición de depresión en niñas de mayor edad. Las niñas además suelen tener baja autoestima y varios problemas emocionales. En ambos sexos, al llegar a la adolescencia, las destrezas de adaptación y cognitivas declinan, lo mismo que el funcionamiento de ejecución, particularmente en lo que atañe a memoria de trabajo, inhibición y planeación (2,18,19).
Epidemiología:
Hay diversas maneras de enfocar la epidemiología del síndrome de X frágil, pero todos los autores concuerdan en la dificultad de obtener la tasa total de prevalencia y por ende la frecuencia génica dentro de una población total. Ha sido más fácil estimar la contribución del síndrome a la etiología del retardo mental y se ha realizado mediante tamizaje de poblaciones de deficientes mentales para detección del marcador cromosómico, con la dificultad de que existen otros marcadores adyacentes a FRAXA en el cromosoma X que pueden producir falsos positivo. El estimado de prevalencia total del síndrome de X frágil obtenido de estudios moleculares en Inglaterra varía de 1:2700 a 1:5700 varones (20) y es de 1:4350 en Australia (1). El análisis molecular ha permitido ahora el estudio de la prevalencia de mujeres portadoras. Un estudio sistemático realizado en mujeres de la población general de Québec encontró que 1: 500 es portadora de un alelo mayor a las 66 repeticiones, y que 1: 500 es portadora de un alelo con un tamaño entre 55 y 63 repeticiones. Estas últimas tienen un riesgo menor de sufrir amplificación del ADN que las primeras, por lo que disminuye la probabilidad de tener un niño afectado (21). Un estudio de incidencia de FRAXA en la población catalana de España, realizado en 5 000 nacimientos consecutivos de varones, encontró una incidencia de varones afectados de 1 en 2 466, de varones portadores de 1 en 1 233, y por deducción, de 1 en 8 333 mujeres afectadas con manifestaciones clínicas y de 1 en 411 mujeres portadoras (22)
El X frágil ha sido detectado en todos los grupos étnicos mayoritarios (8), y se ha observado que un efecto fundador puede ocurrir en algunas poblaciones, por ejemplo la población judía de Túnez presenta una incidencia diez veces mayor que otras poblaciones israelitas, debido a que portan una alta frecuencia de alelos sin la tripleta AGG intercalada (23).
Prevención:
Mientras no se pueda remediar la condición de portador de la mutación, la prevención primaria de la ocurrencia de esta patología se basa en la asesoría genética, adecuada y oportuna, a los miembros de la familia afectada. La prevención basada en el consejo genético es la forma más efectiva de enfrentar este síndrome. En Nueva Gales del Sur, Australia, (con una población de 6,5 millones de habitantes) por ejemplo, la tasa de incidencia ha bajado diez veces, de 4,3/10.000 a 0,5/10.000 como consecuencia del programa de consejo genético, apoyado en diagnóstico prenatal e interrupción del embarazo (24). La prevención secundaria o post-concepción se logra en los países donde se permite la interrupción del embarazo, mediante diagnóstico precoz, embrionario o fetal (25). En cuanto a prevención terciaria, el estudio a fondo de los pacientes con éste síndrome ha permitido plantear estrategias específicas para mejorar su funcionamiento intelectual y su adaptación social (26), de ahí la importancia de la confirmación molecular del diagnóstico clínico.
Tratamiento:
El enfoque más útil en el tratamiento del síndrome X frágil es el multidisciplinario y de conjunto, que debe incluir un maestro de educación especial, un terapeuta de lenguaje, un terapeuta ocupacional, un médico, un psicólogo y un consejero genético (27).
Los niños oportuna y correctamente diagnosticados con el síndrome del cromosoma X frágil se benefician con la adquisición de este conocimiento por parte de sus padres y de sus maestros ya que tienen características particulares que los diferencian de otros niños con retardo o dificultades de aprendizaje de otro origen. Estas peculiaridades hacen necesario un abordaje psicopedagógico especial, el cual ha tenido éxito probado en otros países y ha contribuido al desarrollo de los niños de manera que funcionen con la mayor independencia posible, en ambientes tan normales como es factible (28). Por otro lado, la identificación de personas portadoras en las familias les permite tomar decisiones reproductoras acorde a sus necesidades, circunstancias y valores. Para las personas con alelos estables dentro del ámbito normal de repeticiones CGG, el alivio de su preocupación respecto al riesgo de transmisión es grande.
Situación en Costa Rica:
En el Instituto de Investigaciones en Salud hicimos el primer diagnóstico de FRAXA, tenemos casi 20 años de estudiar el síndrome del cromosoma X frágil (13, 14, 16, 17) y hemos encontrado frecuencias similares a las informadas en muchos otros países. En Costa Rica, no existen datos sobre la inversión económica y social que representa para la sociedad y las familias afectadas, la atención de las personas discapacitadas. Sin embargo los pacientes afectados por FRAXA requieren atención y dedicación de otras personas durante toda su vida. La esperanza de vida de los afectados y portadores es igual que la población general. En casi todos los países del mundo, incluido el nuestro, la mayoría de las personas afectadas por FRAXA no han sido correctamente diagnosticadas y la etiqueta más común en ellos es la de retardo mental de origen oscuro. Como consecuencia, las portadoras y los varones transmisores normales tampoco han sido identificados, de manera que la prevención se hace imposible. Varias razones explican esta deficiencia, principalmente el hecho de que el fenotipo de los afectados, sobre todo los más pequeños, es poco llamativo para los especialistas que tratan esta población, los cuales suelen estar poco informados acerca de esta patología, dado que su conocimiento es de relativa reciente adquisición. Actualmente estamos desarrollando el proyecto "Prevención del síndrome del cromosoma X frágil mediante el tamizaje a poblaciones seleccionadas" y el proyecto "Tamizaje del retardo mental hereditario (FRAXA) en recién nacidos costarricenses" para detectar los probandos (probando es el individuo afectado a través del cual se llama la atención sobre una genealogía), de manera que se abre la posibilidad de estudiar la familia y brindar asesoramiento genético a las personas portadoras.
En el país existe la necesidad de identificar a las familias en las que segrega la mutación FRAXA, pues a diferencia del síndrome de Down, que suele ser un evento esporádico y aislado, el síndrome del cromosoma X frágil es hereditario y a menudo afecta a varios miembros de una misma familia. Por no presentar un fenotipo característico y bien reconocido como en el caso de las trisomías, las personas afectadas suelen explicarse con base a etiologías oscuras o erróneas. No es sino hasta que la biología molecular confirma el diagnóstico de FRAXA, que se identifica correctamente los probandos y por lo tanto a las familias con riesgo, ya que las mutaciones frescas son prácticamente inexistentes. Una vez detectados los probandos, se abre la posibilidad de identificar entre sus familiares, aquellas personas que tienen alto riesgo de engendrar niños similarmente afectados y que se beneficiarían con el asesoramiento genético adecuado y oportuno. Está demostrado que estas acciones contribuyen a disminuir la carga genética que ocasiona FRAXA en la población (24).
El tamizaje del síndrome X frágil:
Tamizaje es el estudio sistemático de personas no diagnosticadas para identificar a los individuos con riesgo alto de padecer una enfermedad, de manera de poder aplicarles un manejo específico el cual no está disponible para toda la gente, ya sea por motivos económicos o de cualquier otra índole. Obviamente la prueba para tamizaje es mucho más sencilla y más barata que la prueba diagnóstica, arroja resultados más pronto, con un grado de especificidad y sensibilidad suficientes, es mínimamente invasiva y tiene buena aceptación por parte de la población diana (29,30).
En 1995, Willemsen describió una prueba adecuada para hacer tamizaje poblacional basada en la ausencia de expresión de FMRP en los linfocitos de los afectados por FRAXA (31,32). Esta es una prueba inmunohistoquímica que se aplica en frotis de sangre periférica e identifica a los afectados que no muestran FMRP en el citoplasma de sus linfocitos (figura 1). Las principales ventajas metodológicas de esta prueba son la rapidez y el estudio directo de la presencia del producto génico. Otra ventaja es el bajo costo relativo a los estudios moleculares. Los resultados han demostrado que la técnica tiene un gran poder de diagnóstico en los varones, en las mujeres es menos específica debido al proceso de inactivación de uno de sus cromosomas X durante la embriogénesis temprana. Así se encontró que algunas mujeres con la mutación tenían porcentajes de expresión de FMRP en los linfocitos que se traslapan con el ámbito de valores de las mujeres control, y además, el grado de retardo mental no se correlacionaba con el porcentaje de expresión de la proteína en este tejido.
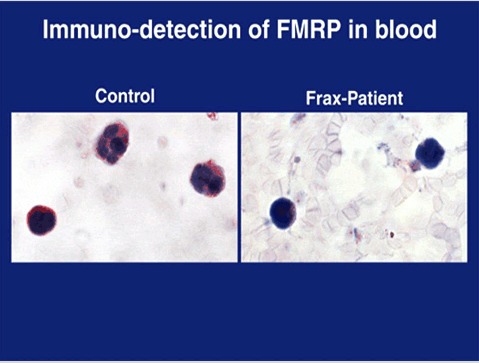
Figura 1. El citoplasma de los linfocitos se tiñe de rojo cuando hay presencia de la proteína FMRP. En los individuos afectados con la mutación no hay proteína y por lo tanto tampoco hay tinción. Fuente: Internet http://www.eur.nl/FGG/CH1/fragx/.
En 1999 este mismo grupo holandés, con el objetivo de mejorar las correlaciones entre los valores de CI y el resultado inmunohistoquímico, tanto en varones como en mujeres, desarrolló la prueba para su aplicación en la raíz del cabello (figura 2). La elección de este tejido obedece a que las células epiteliales y el sistema nervioso derivan de la misma hoja embrionaria, el ectodermo, de manera que la expresión de la proteína en cabellos refleja mejor lo que ocurre en las neuronas (33).
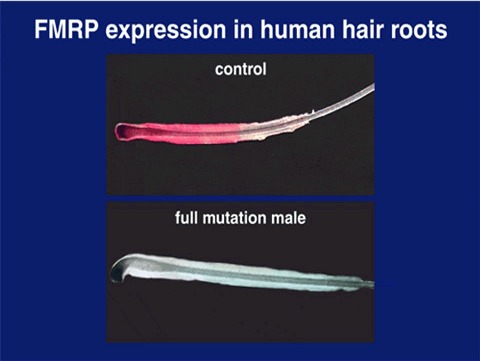
Figura 2. La raíz del cabello se tiñe de rojo cuando hay presencia de la proteína FMRP. En los individuos afectados no hay tinción pues no hay producto génico. Fuente: Internet http://www.eur.nl/FGG/CH1/fragx/.
El proyecto en ejecución "Prevención del síndrome del cromosoma X frágil mediante el tamizaje a poblaciones seleccionadas" está siendo enfocado como un programa de tamizaje para encontrar probandos en escolares con educación especial, como punto de partida para identificar a las familias afectadas para estudio y asesoramiento. Se trata pues de tamizaje tipo cascada cuyo objetivo final es la prevención, sea ésta primaria, secundaria o terciaria. El tamizaje en cascada activo significa buscar los casos en instituciones para personas con retardo mental, autismo o problemas severos de conducta y en las escuelas de enseñanza especial. Para mejorar la eficiencia de este paso se puede pre-seleccionar la población meta de manera de identificar un sub-grupo de personas con mayor probabilidad de resultar tamizaje positivas. Butler y sus colegas encontraron que una manera de clasificar correctamente a cerca del 90% de los varones afectados, es mediante una combinación de historia familiar de problemas de aprendizaje sumado a cinco características clínicas específicas: pliegue plantar, pliegue palmar único, macroorquidismo (después de la pubertad), orejas grandes o prominentes e hiperextensibilidad de las articulaciones (34). Las personas tamizaje positivo posteriormente son objeto de las pruebas confirmatorias, en este caso la hibridación de Southern para detectar la amplificación de la tripleta CGG. Una vez confirmado el caso de FRAXA, se inicia el estudio molecular de más personas afectadas y de portadoras, y el asesoramiento de la familia nuclear y extensa. Para el diagnóstico de las personas portadoras, la técnica que se emplea es la PCR. Se utilizan además plegables y folletos para explicar de manera sencilla lo que estas personas necesitan saber y comprender, para así lograr la prevención buscada. Se mantendrá además el contacto con las familias con el objeto de comunicar nuevos descubrimientos que les sean útiles, tanto para prevención como para tratamiento de los miembros afectados.
La población de estudio consiste de niños, niñas y adolescentes con retardo mental o autismo de origen no precisado o con diagnóstico presuntivo no confirmado (suelen ser dos tercios de los estudiantes en enseñanza especial), alumnos de las escuelas de enseñanza especial del Ministerio de Educación Pública (MEP), de las escuelas privadas o con subvención estatal (una de ellas para adolescentes), del Centro de Atención Integral de Guadalupe, de los Centros Integrados Locales de Rehabilitación en provincias, de los talleres protegidos, de las escuelas para niños de cero a seis años que funcionan en los hospitales R.A. Calderón y Nacional de Niños y del Hospital Neuropsiquiátrico Infantil. Tomando en cuenta datos del Consejo Nacional de Rehabilitación y Educación Especial, se reportan alrededor de 560 casos nuevos de retardo mental por año, de los cuales cerca del 6% son originados por FRAXA. Suponiendo una estadía mínima de cinco años en el sistema escolar, cabe esperar una cantidad de 168 probandos y un número igual de familias, con un promedio de al menos diez miembros estudiados, la mitad de los cuales sería portador o portadora del síndrome, para un total aproximado de 840 personas con riesgo de transmisión de retardo mental beneficiadas con la información que les permite prevenir esta situación.
Se realizan reuniones con los maestros y los padres de familia para informarles acerca de FRAXA y del estudio, ventajas y desventajas de participar en el mismo y otros aspectos de su interés. El tamizaje es voluntario, los padres firman un consentimiento informado para otorgar el permiso para que sus hijos participen en el estudio.
Procedimientos de laboratorio: la prueba de tamizaje es la detección de la expresión de la proteína FMRP en cabellos del sujeto y/o en un frotis de su sangre (un par de gotas obtenidas por punción de la yema del dedo). La elección de una o de otra muestra depende de la colaboración del sujeto.
La detección de la proteína FMRP se realiza mediante las técnicas inmunohistoquímicas desarrolladas por Willemsen y colaboradores en el Departamento de Genética Clínica de La Universidad Erasmus en Rótterdam, en los Países Bajos y puestas a punto en nuestro laboratorio.
La confirmación molecular voluntaria, se realiza en una muestra de sangre de la vena del brazo, anticoagulada con ACD. Las técnicas de extracción del ADN y de análisis directo de la mutación FRAXA son las mismas que usamos de rutina en el laboratorio.
El asesoramiento genético está a cargo de la Unidad de Asesoramiento Genético del INISA, siguiendo las directrices internacionalmente reconocidas (35-37).
A corto plazo informaremos sobre el resultado final de este estudio, las consecuencias a nivel poblacional tendrán que esperar al menos que transcurra una generación para valorar el impacto del tamizaje en cascada en nuestro país.
Referencias:
1. Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet 1996; 64:196-97. [ Links ]
2. Hagerman R. Clinical and diagnostic aspects of fragile X syndrome. En Wells R, Warren ST, Sarmiento M, eds. Genetic instabilities and hereditary neurological diseases. New York: Ed. Academic Press; 1998: 15-25. [ Links ]
3. Verkerk A J M, Pieretti M, Sutcliffe J S, Fu Y H, Kuhl D P, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991; 65:905-914. [ Links ]
4. Oberlé I, Rosseau F, Heitz D, Kretz C, Deveys D et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991; 252:1097-1102. [ Links ]
5. Fu YH, Kuhl DPA, Pizutti A, Pieretti M, Sutcliffe JS, Richards S et al. Variation of the CGG repeat al the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67:1047-1058 [ Links ]
6. Yu S, Pritchard M, Kremer E, Lynch M, Nancarraw J, Baker E, et al. Fragile X genotype characterized by an unstable region of DNA. Science 1991; 252:1179-81. [ Links ]
7. Eichler EE, Holden JJA, Popovich BW, Reiss AL, Snow K, Thibodeau SN, et al. Length of uninterrupted CGG repeats determines stability in the FMR1 gene. Nature Genet 1994; 8: 88-94. [ Links ]
8. Imbert G, Feng Y, Nelson D L, Warren ST, Mandel. 1998. FMR1 and mutations in fragile X syndrome: Molecular biology, biochemistry, and genetics. En Wells R, Warren ST, Sarmiento M, eds. Genetic instabilities and hereditary neurological diseases. New York: Ed. Academic Press. 1998. p. 27-54. [ Links ]
9. Jin P, Warren ST. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet 2000; 9:901-908. [ Links ]
10. Zalfa F, Bagni C. Molecular insights into mental retardation: multiple functions for the fragile X mental retardation protein? Curr Issues Mol Biol 2004; 6:73-88. [ Links ]
11. Siomi H, Ishizuka A, Siomi MC. RNA interference: a new mechanism by which FMRP acts in the normal brain? What can Drosophila teach us? Ment Retard Dev Disabil Res Rev 2004; 10:68-74. [ Links ]
12. Downward J. Science, medicine and the future. RNA interference. BMJ 2004; 328:1245-1248. [ Links ]
13. Castro I, Cuenca P. Tamizaje de sitio frágil en el cromosoma X en una población de retardados mentales. Hallazgos preliminares. Rev Méd Hosp Nal Niños (Costa Rica) 1987; 1:11-14. [ Links ]
14. Castro I, Cuenca P. Frecuencia del síndrome del cromosoma X frágil en la Escuela de Enseñanza Especial "Fernando Centeno Güell" Act Pediatric Costarric 1996; 10:99-106. [ Links ]
15. Maddalena A, Richards C, McGinniss MJ, Brothman AR, Desnick RJ, Grier RE et al. American College of Medical Genetics: Technical standards and guidelines for fragile X. Genet Med 2001; 3:200-205. [ Links ]
16. Cuenca-Berger P, Morales-Montero F, Castro-Volio I. Diagnóstico directo de la mutación que causa el síndrome del cromosoma X frágil. Experiencia en Costa Rica. Acta Med Costarric 2002; 44:27-33. [ Links ]
17. Cuenca-Berger P., Morales-Montero F. & Castro-Volio, I. (2004) Disabilities caused by unstable mutations in Costa Rica. Rev Biol Trop 52 (3): 501-505. [ Links ]
18. Reiss AL, Dant CC. The behavioral neurogenetics of fragile X syndrome: analyzing gene-brain-behavior relationships in child developmental psychopathologies. Develop Psychopathol 2003; 15:927-968. [ Links ]
19. Ferrando-Lucas MT, Banús-Gómez P, López-Pérez G. Aspectos cognitivos en niñas con síndrome X frágil. Rev Neurol 2004; 38 (Supl 1): S53-S57. [ Links ]
20. Morton JE, Bundey S, Webb TP, MacDonald F, Rindl PM, Bullock S. Fragile X syndrome is less common than previously estimated. J Med Genet 1997; 34:1-5. [ Links ]
21. Rousseau F, Rouillard P, Morel M L, Khandjian E W, Morgan K. Prevalence of carriers of premutation.size alleles of the FMR1 gene and implications for the population genetics of the fragile X syndrome. Am J Hum Genet 1995:57: 1006-1018. [ Links ]
22. Rifé M, Badenas C, Mallolas J, Jiménez L, Cervera R, Maya A, et al. Incidence of frágile X in 5000 consecutive newborn males. Genet Test 2003;7:339-43. [ Links ]
23. Falik-Zaccai TC, Shachak E, Yalon M, Lis Z, Borochowitz Z, MacPherson JN, et al. Predisposition to the fragile X syndrome in Jews of Tunisian descent is due to the absence of AGG interruptions on a rare Mediterranean haplotype. Am J Hum Genet 1997; 60:103-112. [ Links ]
24. Turner G, Robinson H, Wake S, Laing S, Partington M. Case finding for the fragile syndrome and its consequences. BMJ 1997; 315: 1223-1226. [ Links ]
25. Sutherland GR, Gedeon A, Korman L, Donnelly A, Byard RW, Mulley JC et al. Prenatal diagnosis of fragile X syndrome by direct detection of unstable DNA sequence. N Engl J Med 1991; 325: 1720-1722. [ Links ]
26. Spiridigliozzi GA, Lachiewicz M, MacMurdo CS, Vizoso AD, ODonnel CM, McConkie-Rossel A, et al. Educating boys with fragile X syndrome. A Guide for parents and professionals. Duke University Medical Center. 1994: 1-20. [ Links ]
27. Hagerman RJ. Medical follow-up and pharmacotherapy. En Hagerman RJ, Cronister A, eds. Fragile X syndrome diagnosis, treatment and research. Baltimore: Ed.John Hopkins; 1996 p. 3-250. [ Links ]
28. Braden M. Fragile, handle with care. Understanding fragile X syndrome. Chapel Hill: Ed. Avanta; 1996. [ Links ]
29. Murray J, Cuckle H, Taylor G, Hewison J. Screening for fragile X syndrome: information needs for health planners. J Med Screen 1997; 4:60-94) . [ Links ]
30. Pembrey ME, Barnicoat AJ, Carmichael B, Bobrow M, Turner G. An assessment of screening strategies for fragile x syndrome in the UK. Health Technol Assess 2001;5 (7). [ Links ]
31. Willemsen R, Mohkamsing S, De Vries B, Devys D, Van den Ouweland A, Mandel JL, et al. Rapid antibody test for Fragile X syndrome. Lancet 1995;345:1147-8. [ Links ]
32. Willemsen R, Smits A, Mohkamsing S,Van Beerendonk H, De Haan A, De Vries B, et al. Rapid antibody test for diagnosing Fragile X syndrome. A validation of the technique. Hum Genet 1997;99:308-11. [ Links ]
33. Willemsen R, Anar B, De Diego Otero Y, De Vries BBA, Hilhorst-Hofstee Y, Smits A, et al. Noninvasive test for Fragile x syndrome using hair root analysis. Am J Hum Genet 1999;65:98-103. [ Links ]
34. Butler MG, Mangrum T, Gupta R, Singh DN. A 15-item checklist for screening mentally retarded males for the fragile X syndrome. Clin Genet 1991; 39:347-54. [ Links ]
35. McIntosh N, Gane LW, McConkie-Rosell A, Bennett RL. Genetic counseling for fragile X syndrome: recommendations of the National Society of Genetic Counselors. J Genet Couns 2000; 9:303-25. [ Links ]
36. Carrasco M. La comunicación del diagnóstico a las familias afectadas por el síndrome X frágil. Rev Neurol 2001; 33 (Supl 1):S37-S41. [ Links ]
37. Gane LW, Cronister A. Genetic counseling. En: Hagerman RJ, Hagerman PJ, eds. Fragile X syndrome: Diagnosis, Treatment and Research. 3rd ed. Baltimore: Johns Hopkins University Press; 2002:251-286. [ Links ]
1. Instituto de Investigaciones en Salud (INISA), Universidad de Costa Rica.
*Correspondencia: Dra. Isabel Castro, INISA, Universidad de Costa Rica, 2060 San Pedro, San José, Costa Rica; tel: (506) 207-3293; fax: (506) 207-5130; correo electrónico: icastro@cariari.ucr.ac.cr
