











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
PermalinkIntroducción
La correcta determinación de las poblaciones e individuos bajo la categoría taxonómica de especie es fundamental en la resolución de preguntas sobre la historia evolutiva, ecología, diversidad funcional, e impactos antrópicos sobre la distribución y diversidad de los organismos (Roy, Girman, Taylor, & Wayne, 1994; Cameron, Hines, & Williams, 2007; Hines, 2008; Condamine & Hines, 2015). Sin embargo, es de resaltar que una de las grandes limitantes para los estudios que conducen a responder las preguntas arriba mencionadas, son las cortas escalas temporales a las cuales se puede generar información empleando material recolectado de manera reciente, así como la baja cobertura a nivel geográfica de los estudios (Wandeler, Hoeck, & Keller, 2007). Es así como las colecciones biológicas revisten gran importancia como fuente de información histórica, geográfica y ecológica de los organismos, muchos de ellos especímenes raros o capturados en áreas de difícil acceso, o incluso que por cambios antrópicos, desaparecieron (Gilbert, Moore, Melchior, & Worobey, 2007; Wandeler et al., 2007). Las colecciones no solo constituyen un repositorio de datos morfológicos de los especímenes allí preservados, sino también de la información genética en cada uno de ellos (Mitchell, 2015). A través de la integración de la información morfológica y molecular que proveen las colecciones biológicas es posible la resolución de complejos de especies (Hausmann et al., 2009), detectar la aparición de rasgos genéticos y/o morfológicos que confieren ventajas evolutivas (ej. resistencia a insecticidas) y determinar diversidad y distribución histórica de poblaciones, y de este modo evaluar el efecto de variables como el cambio climático o la actividad antrópica sobre dichas especies (Wandeler et al., 2007).
No obstante lo anterior, el potencial de las colecciones biológicas como proveedoras de información genética de los individuos allí preservados ha sido subutilizada debido a la dificultad en la extracción de material genético de calidad de los especímenes (Mitchell, 2015; Sproul & Maddison, 2017). A partir de la toma de una muestra de un espécimen, o la muerte del mismo, inicia un proceso de degradación del ADN, que dificulta su uso apropiado en estudios genéticos (Zimmermann et al., 2008). Investigaciones empleando muestras depositadas en colecciones biológicas han tenido resultados variables en cuanto al éxito en la amplificación de marcadores moleculares a partir de estos tejidos (Hausmann et al., 2009; Hebert, et al., 2013; Mitchell, 2015), que se ve influenciado por el tamaño de la muestra almacenada, el medio de preservación empleado, la edad del tejido o los métodos empleados para la eliminación de plagas al interior de la colección (Ackery, Testa, Ready, Doyle, & Pinniger, 2004; Bisanti, Ganassi, & Mandrioli, 2009; Hebert et al., 2013). Particularmente en el caso de los insectos montados en alfileres entomológicos, en seco sin ningún medio adicional de preservación, el ADN se deteriora en un lapso de pocos años siendo adecuado para estudios moleculares por un periodo de entre 10 a 15 años posterior al depósito del espécimen en la colección (Hernández-Triana et al., 2014), periodo a partir del cual el éxito de amplificación se reduce drásticamente (Zimmermann et al., 2008). De este modo y teniendo en cuenta las limitaciones del uso de muestras de colección en la obtención de información genética, recientes estudios proponen la implementación de técnicas de secuenciación de nueva generación (NGS por sus siglas en inglés) (Sproul & Maddison, 2017), como la piro- secuenciación (Shokralla et al., 2011), así como diferentes modificaciones de los protocolos de PCR (Mitchell, 2015). Esto último, ha permitido la obtención de fragmentos cortos de ADN conocidos como mini-barcodes, que pueden ser ensamblados para la obtención de secuencias del código de barras completas (p. ej. Strutzenberger, Brehm, & Fiedler, 2012; Françoso & Arias, 2013; Hebert et al., 2013), o empleadas de manera independiente en la determinación molecular de especímenes con alta degradación del material genético (ver Hajibabaei et al., 2006, Hernández-Triana et al., 2014).
En la actualidad, los polinizadores se encuentran en estado de amenaza a nivel mundial debido a la intensificación de la agricultura, el uso de pesticidas, el cambio climático, la destrucción de hábitat y la introducción de especies y patógenos exóticos (Potts et al., 2010). Particularmente para las abejas del género Bombus, en la actualidad se registra la pérdida de biodiversidad más grande en su historia evolutiva (Condamine & Hines, 2015). Este género cuenta con cerca de 265 especies reconocidas, que ocupan una gran diversidad de hábitats, desde praderas alpinas en el viejo mundo, hasta bosques tropicales (Williams & Jepsen, 2017).
En Colombia se registran nueve especies del género, que ocupan todos pisos altitudinales desde el nivel del mar hasta los 4 800 metros sobre el nivel del mar (m.s.n.m.) (Nates, 2004), sin embargo, existe una distribución diferencial de estas especies: Las de cara corta (B. funebris, B. hortulanus, B. robustusB. rubicundus, y B. melaleucus) con excepción de B. melaleucus, son las más comunes en ecosistemas montañosos (a partir de los 2 100 m.s.n.m) y las especies de cara larga (B. atratus, B. pullatus, B. excellens y B. transversalis) presentes exclusivamente en tierras bajas (B. transversalis) o con un amplio rango altitudinal, aunque más frecuentes hacia la tierras bajas (B. atratus, B. pullatus, B. excellens) (Liévano, Ospina, & Nates, 1991). Las especies de cara corta residentes en alta montaña, así como B. atratus se constituyen como especies modelo para el estudio de la biología de estos organismos dada su abundancia y amplia distribución, mientras que B. melaleucus, y B. excellens son muy poco frecuentes (Osorno & Osorno, 1938; Pinilla-Gallego, Ospina, & Cure, 2017). A nivel de conservación, hasta el momento no existe un estimativo de la fluctuación de las poblaciones de estas especies. Sin embargo, a partir de registros realizados en el marco de los diferentes inventarios regionales se observa la desaparición de especies de abejorros de lugares donde estaban previamente reportados (Osorno & Osorno, 1938; Pinilla-Gallego et al., 2017), así como la contracción de las áreas de distribución. Una de las grandes dificultades para el desarrollo de estudios encaminados a determinar el estado de las poblaciones de abejorros, así como de otras especies silvestres de abejas en Colombia es el desconocimiento de la diversidad (Pinilla-Gallego et al., 2017), para el caso particular de Bombus la diversidad intra-especifica. Estudios realizados en Europa (ej. Lecocq et al., 2015; Lecocq et al., 2016a), en Asia (ej. Williams et al., 2012) y en algunos países de Sur América (ej. Françoso, Zuntini, Carnaval, & Arias, 2016), resaltan la importancia del conocimiento de la diversidad genética de las especies de Bombus en la determinación de las áreas reales de distribución, el estado de las poblaciones (Françoso, Zuntini, & Arias, 2019) y consecuentemente, en la formulación de políticas de manejo y protección de estas especies. Teniendo en cuenta lo anterior, el objetivo de este estudio fue identificar y obtener una secuencia mini-barcode para uso en determinación taxonómica y estimación de relaciones filogenéticas de especies del género Bombus, usando muestras de tejido (musculo y/o extremidad derecha del mesotórax) de ejemplares depositados en la colección biológica Laboratorio de Investigaciones en Abejas de la Universidad Nacional de Colombia (LABUN) a partir de año 1980, en el marco de un proyecto de caracterización genética de abejorros en su distribución natural al interior de Colombia.
Materiales y métodos
Especímenes analizados: En el presente estudio se obtuvo información genética de dos grupos de muestras del género Bombus; el primer grupo se compone de especímenes de las especies B. atratus, B. funebris, B. hortulanus, B. robustus y B. rubicundus montados en alfiler entomológico previo sacrificio en cámara letal con Cianuro de Potasio, y preservados en seco en LABUN (en adelante muestras de museo), en un periodo de entre 7 a 38 años de antigüedad, recolectadas en las tres cordilleras de Colombia así como en los llanos Orientales (Tabla 1). El segundo grupo consiste en 31 muestras de las especies arriba mencionadas, recientemente recolectadas y de las que se obtuvieron secuencias del marcador COI. Los ejemplares provienen de la Cordillera Oriental (Departamento de Cundinamarca), Cordillera Central (departamento de Risaralda) y la cordillera occidental (departamentos de Risaralda y Valle del Cauca). Las características de los ecosistemas y ubicaciones geográficas de las localidades de muestreo se muestran en el Apéndice Digital 1.
TABLA 1 TABLE 1 Secuencias obtenidas en el estudio. Se provee el número identificador de colección LABUN, la localidad de muestreo, la fecha de recolecta y el código GenBank de las secuencias obtenidas Sequences obtained in the study. The LABUN collection identification number, the sampling localities, the collection date and the GenBank code of the sequences obtained are provided
| Especie | N° de Colección | Localidad | Cord. | Fecha | Tamaño amplificado | Positivos a partir de pata | Positivos a partir de músculo | N° GenBank |
| Bombus atratus | 13* | Cauca. Inza Vda Tierras Blancas, 2 200 msnm | Central | 1982 | 406 pb | - | ATRA83,4 | MK544970 |
| Bombus hortulanus | 29* | Cauca. Inza Vda Tierras Blancas, 2 200 msnm | Central | 1982 | 161 pb | - | X8 | |
| Bombus rubicundus | 81 | Cundinamarca. Páramo San Cayetano, 3 650-3 690 msnm | Oriental | 1983 | 108 pb | RUBI98 | MK544971 | |
| Bombus atratus | 690* | Cundinamarca. Cabrera. Vía Peñas blancas, 1 950 msnm | Oriental | 1983 | 98pb | - | ATRA88 | MK544972 |
| Bombus hortulanus | 1 045 | Cundinamarca. Subachoque, 2 600 msnm | Oriental | 1985 | 364pb | ROB55,8 | MK544973 | |
| Bombus atratus | 1 111* | Cundinamarca. Subachoque, 2 600 msnm | Oriental | 1985 | 369pb | X5 | ||
| Bombus atratus | 1 112* | Cundinamarca. Subachoque, 2 600 msnm | Oriental | 1985 | 137pb | ATRA28 | MK544974 | |
| Bombus funebris | 1 179 | Cundinamarca. Páramo de Chizacá Laguna Negra, 3 780msnm | Oriental | 1985 | 354 pb | |||
| Bombus hortulanus | 2 039* | Magdalena. Vía San Lorenzo, 1 900-2 000 msnm | Oriental | 1987 | 357 pb | HOR25 | MK544975 | |
| Bombus hortulanus | 15 805* | Risaralda. PNN Tatamá, 3 579 msnm. | Occidental | 1993 | 339pb | X5 | ||
| Bombus hortulanus | 15 810* | Risaralda. PNN Tatamá, 3 579 msnm. | Occidental | 1993 | 365pb | HOR25 | MK544977 | |
| Bombus funebris | 15 823 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2001 | 374pb | FUN15 | MK544978 | |
| Bombus funebris | 15 824 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2001 | 225pb | FUN85 | MK544979 | |
| Bombus funebris | 15 826 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2001 | 407pb | FUN77 | MK544980 | |
| Bombus rubicundus | 15 837 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2001 | 407pb | RUBI107 | MK544981 | |
| Bombus rubicundus | 15 838 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2001 | 407pb | RUBI97 | MK544982 | |
| Bombus rubicundus | 15 839 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2001 | 407pb | RUBI117 | MK544983 | |
| Bombus robustus | 18094 | Santander. Floridablanca. Casianoalto, 1 700 msnm | Oriental | 2004 | 407pb | ROB47 | MK544984 | |
| Bombus hortulanus | 18 095* | Santander. Floridablanca. Casianoalto, 1 700 msnm | Oriental | 2004 | 407pb | HOR27 | MK544985 | |
| Bombus hortulanus | 18 096* | Santander. Floridablanca. Casianoalto, 1 700 msnm | Oriental | 2004 | 407pb | HOR27 | MK544986 | |
| Bombus robustus | 21 227 | Nariño. Pasto, 2 617 msnm | Occidental | 1999 | 406pb | ROB27 | MK544987 | |
| Bombus hortulanus | 26 473* | Antioquia. Envigado. Loma del escobero, 2 459 msnm | Central | 2010 | 407pb | HOR27 | MK544988 | |
| Bombus hortulanus | 26 474* | Antioquia. Envigado. Loma del escobero, 2 459 msnm | Central | 2010 | 407pb | HOR27 | MK544989 | |
| Bombus hortulanus | 26 476* | Antioquia. Envigado. Loma del escobero, 2 459 msnm | Central | 2010 | 407pb | HOR27 | MK544990 | |
| Bombus atratus | 26 499* | Antioquia. Carmen de Viboral, 2 110 msnm | Central | 2009 | 485pb | ATRA14 | ATRA13 | MK544991 |
| Bombus atratus | 26 502* | Antioquia. Carmen de Viboral, 2 110 msnm | Central | 2009 | 658pb | ATRA16,7 | MK544992 | |
| Bombus atratus | 26 503* | Antioquia. Carmen de Viboral, 2 110 msnm | Central | 2009 | 407pb | ATRA77 | MK544993 | |
| Bombus robustus | 27 930 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2016 | 658pb | ROB11,2 | ||
| Bombus robustus | 27 931 | Páramo El Duende. Valle del Cauca, 2 682 msnm | Occidental | 2016 | 658 pb | ROB21,2 | MK531257 | |
| Bombus hortulanus | 27 932 | Páramo El Duende. Valle del Cauca, 2 682 msnm | Occidental | 2016 | 658 pb | HOR11,2 | MK531278 | |
| Bombus funebris | 28 110 | Cundinamarca. Choachí. Mata Redonda, 3 400 msnm | Oriental | 2016 | 658 pb | FUN11,2 | MK531297 | |
| Bombus funebris | 28 111 | Cundinamarca. Choachí. Mata Redonda, 3 400 msnm | Oriental | 2016 | 658 pb | FUN21,2 | MK531298 | |
| Bombus funebris | 28 112 | Cundinamarca. Choachí. Mata Redonda, 3 400 msnm | Oriental | 2016 | 658 pb | FUN31,2 | MK531299 | |
| Bombus hortulanus | 28 126 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2016 | 658 pb | HOR21,2 | MK531279 | |
| Bombus hortulanus | 28 142 | Cundinamarca. Simijaca, 2 567 msnm | Oriental | 2015 | 658 pb | HOR31,2 | MK531282 | |
| Bombus robustus | 28 143 | Cundinamarca. PNN Chingaza, 2 900-3 100 msnm | Oriental | 2016 | 658 pb | ROB31,2 | MK516126 | |
| Bombus robustus | 28153 | Cundinamarca. Salto de Tequendama, 2 260 msnm. | Oriental | 2017 | 658 pb | ROB41,2 | MK516134 | |
| Bombus robustus | 28 154 | Cundinamarca. Fómeque. La Laja, 3 200 msnm | Oriental | 2017 | 658 pb | ROB51,2 | MK516132 | |
| Bombus robustus | 28 156 | Cundinamarca. Fómeque. La Laja, 3 200 msnm | Oriental | 2017 | 658 pb | ROB61,2 | MK516133 | |
| Bombus rubicundus | 28 165 | Risaralda. PNN Tatamá, 3 579 msnm. | Occidental | 2017 | 658 pb | RUBI11,2 | MK516166 | |
| Bombus rubicundus | 28 166 | Risaralda. PNN Tatamá, 3 579 msnm | Occidental | 2017 | 658 pb | RUBI21,2 | MK516167 | |
| Bombus rubicundus | 28 167 | Risaralda. PNN Tatamá, 3 579 msnm | Occidental | 2017 | 658 pb | RUBI31,2 | MK516168 | |
| Bombus rubicundus | 28 169 | Risaralda. PNN Tatamá, 3 579 msnm | Occidental | 2017 | 658 pb | RUBI41,2 | MK516170 | |
| Bombus robustus | 28 177 | Risaralda. PNN Tatamá, 3 579 msnm. | Occidental | 2017 | 658 pb | ROB71,2 | MK516138 | |
| Bombus rubicundus | 28 183 | Risaralda. PNN Tatamá, 3 579 msnm. | Occidental | 2017 | 658 pb | RUBI51,2 | MK516174 | |
| Bombus atratus | 28 191 | Risaralda. Planes de San Rafael, 2 096 msnm | Occidental | 2017 | 658 pb | ATRA21,2 | MH221875 | |
| Bombus atratus | 28 194 | Risaralda. Planes de San Rafael, 2 096 msnm | Occidental | 2017 | 659 pb | ATRA31,2 | MH221876 | |
| Bombus rubicundus | 28 339 | Risaralda. PNN Los Nevados. EL Jordan, 3 300 msnm | Central | 2017 | 660 pb | RUBI61,2 | MK531294 | |
| Bombus rubicundus | 28 340 | Risaralda. PNN Los Nevados. EL Jordan, 3 300 msnm | Central | 2017 | 661 pb | RUBI71,2 | MK531296 | |
| Bombus rubicundus | 28 342 | Risaralda. PNN Los Nevados. El Jordán, 3 300 msnm | Central | 2017 | 663 pb | RUBI81,2 | MK531296 | |
| Bombus hortulanus | 28 343 | Risaralda. PNN Los Nevados. Agua bonita, 3 200 msnm | Central | 2017 | 664 pb | HOR31,2 | MK531285 | |
| Bombus robustus | 28 348 | Risaralda. PNN Los Nevados. El Jordan, 3 300 msnm | Central | 2017 | 665 pb | ROB81,2 | MK516146 | |
| Bombus funebris | 28 363 | Risaralda. PNN Los Nevados. El Jordan, 3 300 msnm | Central | 2017 | 666 pb | FUN41,2 | MK531305 | |
| Bombus funebris | 28 364 | Risaralda. PNN Los Nevados. El Jordan, 3 300 msnm | Central | 2017 | 667 pb | FUN51,2 | MK531306 | |
| Bombus funebris | 28 366 | Risaralda. PNN Los Nevados. El Jordan, 3 300 msnm | Central | 2017 | 668 pb | FUN61,2 | MK531308 | |
| Bombus atratus | MON1 | Cundinamarca. Mondoñedo, 2 540 msnm | Oriental | 2015 | 669 pb | ATRA61,2 | MH221874 | |
| Bombus atratus | BV3 | Cundinamarca. Simijaca, 2 567 msnm | Oriental | 2015 | 670 pb | ATRA41,2 | MH221872 | |
| Bombus atratus | BV5 | Cundinamarca. Simijaca, 2 567 msnm | Oriental | 2015 | 671 pb | ATRA51,2 | MH221873 |
Se indica el tipo de muestra (extremidad derecha del mesotórax o tejido), a partir de la cual se realizó la amplificación: *: Muestras de las que se realizó extracción de ADN a partir de extremidad y músculo. N/A: no aplica. - no amplifica. Los superíndices en cada uno de los haplotipos indican la combinación de primers con las que se obtuvo una amplificación: 1: BarbeeF/ MtD9, 2: LCO1490 / HC02198, 3: BarbeeF/2248R, 4: 2166F/2386R, 5:2338F/MtD9, 6: LCO1490/MLepR3, 7: MLepF1/HC02198, 8: MLepF1/MLepR3. Por cada individuo se establece el nombre del haplotipo obtenido.
The type of sample (the right limb of the mesothorax or tissue), from which the amplification was performed is indicated: *: Samples from which DNA was extracted from the limb and muscle. N / A: Not apply. - : no amplification was obtained. The name of the haplotype obtained is established for each individual. The superscripts in each of the haplotypes indicate the combination of primers with which an amplification was obtained: 1: BarbeeF / MtD9, 2: LCO1490 / HC02198, 3: BarbeeF / 2248R, 4: 2166F / 2386R, 5: 2338F / MtD9, 6: LCO1490 / MLepR3, 7: MLepF1 / HC02198, 8: MLepF1 / MLepR3.
Extracción de ADN, amplificación de Citocromo Oxidasa I (COI) y secuenciación: Para cada uno de los especímenes de museo a los que previamente se le realizó la determinación morfológica, se removió la extremidad derecha del mesotórax cortándola por debajo de la coxa usando tijeras para micro disección. Para la toma de muestras entre especímenes las tijeras se esterilizaron por medio de calor y alcohol absoluto. Posteriormente, 45 especímenes de las especies Bombus atratus (27), y B. hortulanus (18) fueron puestos en cámara húmeda durante 24 horas, para luego realizar la extracción de una muestra de musculo torácico. Las extremidades extraídas se seccionaron en tres partes para permitir la penetración eficiente del buffer de extracción y la proteinasa K mientras que las muestras de tejido muscular se procesaron sin maceración para evitar el daño mecánico del material genético (Mitchell, 2015).
En cuanto a los especímenes recientemente recolectados, tras la determinación morfológica realizada siguiendo los caracteres propuestos por Liévano, Ospina y Nates (1994), se obtuvieron muestras de músculo torácico para los análisis moleculares. Los implementos como pinzas, agujas y tijeras de micro disección empleadas en la toma de muestras se esterilizaron entre individuos, primero siendo flameados y luego sumergidos en etanol absoluto para evitar la contaminación con ADN ajeno a la muestra. Todas las extracciones de ADN se realizaron empleado el kit DNeasy Blood & Tissue (Quiagen). Tanto las muestras frescas como las muestras de museo se incubaron durante 12 horas a 56 °C, sin embargo, a las muestras de museo no se les aplicó agitación. Adicionalmente, solo las muestras de musculo provenientes de especímenes frescos se trituraron manualmente con maceradores desechables.
Posteriormente, en el LABUN y en el laboratorio del Grupo de estudio Relación Parásito Hospedero (GERPH), se llevó a cabo la estandarización de las reacciones en cadena de la polimerasa (en adelante, PCR por sus siglas en inglés) para la obtención de fragmentos de COI a partir de la combinación de diferentes primers sugeridos por Folmer et al., (1994), Hajibabaei et al., (2006), Françoso & Arias (2013) y Hernández-Triana et al., (2014) (Tabla 2).
TABLA 2 TABLE 2 Primers y combinaciones empleadas en este estudio Primers and combinations used in this study
| Combinación de primers | Secuencia | Autor | Posición en fragmento amplificado de Bombus | Tamaño del fragmento (pb) |
| BarbeeF/ MtD9 | BarbeeF: 5’-CAA CAA ATC ATA AAA ATA TTG G-3’ | (Françoso & Arias, 2013), (Simon et al., 1994) | 1-658 | 658 |
| BarbeeF/2248R | 2248R: 5’-CAA AAT CTA ATA TTA TTT ATT CG-3’ | 1-231 | 231 | |
| 2338F: 5’-CAT ATT TAT ATC ATT CAT CTC C-3’ | ||||
| 2166F/2386R | 2166F: 5’-GGA GGA TTT GGT AAT TTT TTA ATT CC-3’ | 204-380 | 176 | |
| 2386R: 5’-GAA AAA ATT GTA AAA TCA AC-3’ | ||||
| 2338F/MtD9 | MtD9: 5’-CCC GGT AAA ATT AAA ATA TAA ACT TC-3’ | 375-658 | 283 | |
| LCO1490/HC02198 | LCO1490:5’-GGT CAA CAA ATC ATA AAG ATA TTGG-3’ | (Folmer et al., 1994) | 1-658 | 658 |
| MLepF1/HC02198 | HC02198: 5’-TAAACTTCAGGGTGACCAAAAAATCA-3’ | 252-658 | 407 | |
| MLepR3/LCO1490 | MLepF1: 5’-GCT TTC CCA CGA ATA AAT AAT A-3’ | (Hajibabaei et al., 2006) | 1-404 | 404 |
| MLepF1/MLepR3 | MLepR3: 5’-GCT AAR TGW ARD GAA AAA ATW GC-3’ | (Hernández-Triana et al., 2014) | 252-404 | 152 |
Se indica la posición del fragmento amplificado en la secuencia código de barras para el género Bombus. pb: pares de bases.
The position of the amplified fragment in the barcode sequence for the genus Bombus is indicated. bp: base pair.
De este modo, con las combinaciones de los primers de Folmer et al. (1994), Hajibabaei et al. (2006) y Hernández-Triana et al. (2014) MLEPFI/ HCO2198 y LCO1490/MLEPR3 se obtuvieron dos fragmentos de cerca de 400 pb (pares de bases), mientras que con la combinación MLEPF1/MLEPR3 se obtuvo un fragmento de 152 pb. Adicionalmente, con las combinaciones de los primers de BarbeeF/2248R se obtuvo un fragmento de 231 pb, de la combinación 2166F/2386R se obtuvo el fragmento de 176 bp y de la combinación 2338F/MtD9 un fragmento de 283 bp (Simon et al., 1994; Françoso & Arias, 2013). Adicionalmente con las combinaciones BarbeeF/ MtD9 (Simon et al., 1994; Françoso & Arias, 2013) y LCO1490/HCO2198 (Folmer et al., 1994), se obtuvieron las secuencias de código de barras de 658 pb para los especímenes recientemente recolectados (Tabla 2). Los productos de amplificación se visualizaron en geles de agarosa al 1.5 % y se purificaron usando el protocolo de precipitación diferencial con Acetato de Amonio (Bensch et al., 2000). Finalmente, estos productos se secuenciaron en ambos sentidos usando un equipo 3730 xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
Ensamblaje de datos, análisis filogenéticos y divergencia genética: Los fragmentos amplificados se editaron y ensamblaron con MEGA v7.0 (Kumar, Stecher, & Tamura, 2016) y alineados con las secuencias obtenidas a partir de las muestras de tejido de los especímenes recientemente recolectados empleando ClustalW (Thompson, Gibson, & Higgins, 2003).
Una vez obtenidas las secuencias se construyeron siete alineamientos de acuerdo con los tamaños de los fragmentos obtenidos con las diferentes combinaciones de primers analizadas, a partir de los cuales se realizaron las reconstrucciones filogenéticas empleando inferencia bayesiana en el programa Mr. Bayes v3.2 (Ronquist et al., 2012). Así, con el objeto de determinar la consistencia de la información obtenida a partir de especímenes de museo, con respecto a las secuencias obtenidas a partir de ejemplares recientes, se construyó un alineamiento de un fragmento final de 658 pb, que incluyó las secuencias provenientes de los ejemplares recientemente recolectados y los fragmentos de diferentes longitudes ensamblados a partir de mini-barcodes obtenidos a partir de especímenes de museo (Apéndice Digital 2). El modelo de sustitución sugerido por Jmodeltest (Darriba, Taboada, Doallo, & Posada, 2012) fue GTR+I +G. Dos análisis independientes se llevaron a cabo empleando seis cadenas de MCMC, 5 millones de generaciones, y realizando muestreos cada 100 generaciones. La convergencia del análisis se determinó por el valor de la diferencia entre las desviaciones estándar medias menor de 0.01. Después de descartar el 25 % de los árboles, las topologías se obtuvieron sobre un total de 75 000 árboles sobre los que se generó un consenso de 50 %. Las filogenias obtenidas se editaron en el programa FigTree v1.3.1 (Rambaut & Drummond, 2010).
Adicionalmente se calcularon las distancias genéticas intra e inter-especificas empleando el modelo de sustitución Kimura 2 parámetros (K2P), implementado en el software MEGA v7.0 (Kumar et al., 2016). Finalmente, con el fin de evaluar con un mayor detalle las relaciones genéticas entre los diferentes haplotipos del COI de los especímenes analizados, se calcularon redes de haplotipos para cada uno de los fragmentos por medio del software TCS (Clement, Posada, & Crandall, 2000).
Con el objeto de determinar cuál de los fragmentos amplificados puede ser empleado en la reconstrucción de la relaciones filogenéticas de las especies analizadas, así como puede ser usado de manera eficiente como mini-barcode, se construyeron los restantes seis alineamientos, de acuerdo con los fragmentos amplificados por las diferentes combinaciones de primers arriba mencionadas, incluyendo un número fijo de 31 taxones correspondientes a fragmentos obtenidos de especímenes recientemente recolectados, y de 1 a 23 secuencias amplificadas a partir de material de museo. Es así como en el segundo alineamiento en el que se incluyeron secuencias de 231 pb obtenidas con los primers BarbeeF/2248R, se incluyeron tres secuencias de la especie B. atratus amplificadas de ejemplares de museo. Para el tercer alineamiento, que abarca desde la posición 220 hasta la posición 446 del fragmento de 658 pb, amplificada con la combinación 2166F/2386R, se emplearon seissecuencias de B. atratus, una secuencia de B. funebris, cinco secuencias de B. hortulanus, dos secuencias de B. robustus y cuatro secuencias de B. rubicundus. En el cuarto alineamiento, en el que se analizaron las secuencias amplificadas con la combinación 2338F/MtD9, se incluyeron 18 secuencias provenientes de museo: dos de B. atratus, tres correspondientes a B. funebris, dos secuencias de B. hortulanus, ocho de B. robustus y tres de B. rubicundus. En el quinto alineamiento que incluye secuencias de 293 pb obtenidas con los primers LCO1490/MLEPR3, se emplearon el mismo número de secuencias como fue establecido para la construcción del segundo alineamiento.
En el sexto alineamiento se incluyeron 17 secuencias de museo amplificadas con la combinación MLEPFI/ HCO2198, dos de B. atratus, dos de B. funebris, ocho de B. hortulanus, dos de B. robustus y 3 de B. rubicundus, mientras que, para el séptimo, en el que se usó la combinación MLEPF1/MLEPR3 se emplearon cinco secuencias de B. atratus, cinco de B. funebris, 8 de B. hortulanus, dos de B. robustus y cuatro de B. rubicundus (Apéndice Digital 2).
Para los análisis filogenéticos realizados con los seis alineamientos construidos a partir de los fragmentos de 407, 331, 231, 176 y 152 pb., se realizaron dos corridas independientes en las que se emplearon seis cadenas de MCMC, con 3X106 generaciones, sobre las que se realizó un muestreo cada 100 generaciones. La diferencia entre las desviaciones estándar medias fue menor de 0.01.De este modo, posterior a la eliminación del 25 % de los árboles, las topologías se calcularon sobre 45 000 árboles, a los que se aplicó un consenso de 50 %.
Resultados
Calidad de ADN obtenido yéxito de amplificación: De un total de 96 muestras de museo analizadas, se extrajo ADN que se logró amplificar en 34 (35.4 %). De estas muestras se obtuvo un producto de amplificación secuenciable de 27 (28.1 %), dentro de las que predominaron las secuencias de entre 300 a 407 pb (Fig. 1A), amplificadas hacia el extremo 3’ del fragmento de 658 pb usando las combinaciones de primers MLEPFI/ HCO2198 y 2338F/MtD9 (Fig. 1B). Aunque en general las mayores concentraciones e integridades se obtuvieron en las extracciones realizadas a partir de la extremidad derecha del mesotórax, de las muestras obtenidas a partir de especímenes de entre 33 y 38 años de edad de almacenamiento al momento de la extracción, se observó un mayor éxito de amplificación con músculo torácico (6.06 vs. 4.54 % obtenido de extremidades). Sin embargo, en las muestras comprendidas en dicho lapso de tiempo, el éxito de amplificación fue bajo (7.6 %, cinco de 66 muestras), mientras que las muestras recolectadas y almacenadas a partir de 1987, presentaron una mayor eficiencia en la extracción de ADN a partir de extremidad, así como una mayor proporción de muestras con ADN amplificable (63.3 % de 30 muestras); obteniéndose un mayor éxito de amplificación en muestras depositadas a partir del 2001 (Fig. 1C).
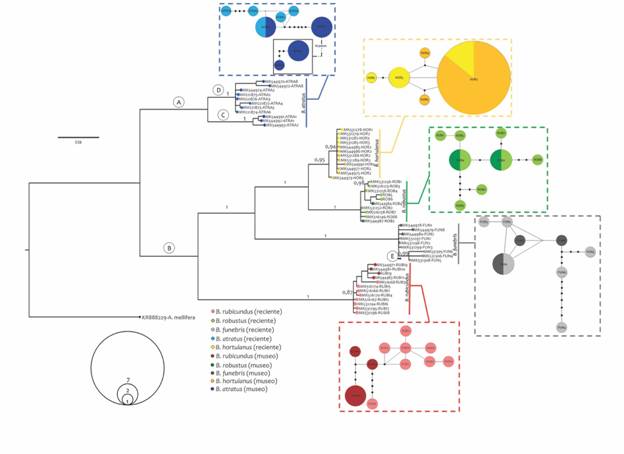
Fig. 1 Fig. 1 Número de muestras a partir de las cuales fue posible extraer ADN y realizar amplificaciones con diferentes primers sobre el material obtenido en muestras de museo con diferentes edades, desde 1980 hasta 2010. A. Longitud de fragmentos y cantidad de muestras para las que se obtuvo cada fragmento, B. cantidad de muestras amplificadas empleando cada una de las combinaciones de oligonucleótidos propuestas en el estudio (Tabla 1). C. numero de muestras de las que se extrajo ADN a partir de musculo torácico o extremidad media del mesotórax. Number of samples from which it was possible to extract DNA and make amplifications with different primers using the material obtained in museum samples with different ages, from 1980 to 2010. A. Length of fragments and number of samples with successful amplification for each fragment, B. number of samples amplified using each of the oligonucleotide combinations proposed in the study (Table 1). C. number of samples from which DNA was extracted from thoracic muscle or middle limb of the mesothorax.
Relaciones filogenéticas y divergencia genética: Al comparar las secuencias obtenidas de especímenes de museo, con secuencias de la misma especie amplificadas a partir de material recolectado recientemente, se observaron varias sustituciones en las secuencias de museo. Dichas sustituciones derivaron en un incremento en el valor de la distancia genética intraespecie (Tabla 3), que para el caso de B. atratus, alcanza un valor superior al 3.7 % reportado por Júnior, Santos y Silveira (2015), para la diferenciación de especies con un fragmento similar (471 pb). Sin embargo, en ningún caso las distancias genéticas intraespecíficas superaron las interespecíficas en las especies analizadas (Tabla 3 vs. Tabla 4). Adicionalmente se observó que la presencia de polimorfismos o posiciones ambiguas, así como la amplificación de fragmentos cortos de baja calidad incrementa en muestras con una edad superior a los 30 años (p. e. especímenes 29, 1179 y 1111) (Tabla 1, Apéndice Digital 2).
TABLA 3 TABLE 3 Distancias genéticas intraespecificas (Dist.) expresadas en porcentaje y desviación standard (SD) calculadas sobre muestras recientemente recolectadas, así como muestras recientemente recolectadas y ejemplares de museo, usando un fragmento de 658 pb del gen COI Intraspecific genetic distances (Dist.) expressed in percentage and standard deviation (SD) calculated on newly collected samples, as well as recently collected samples plus museum specimens, using a 658 bp fragment of the COI gene
| Especie | Recolecta reciente | Museo + recolecta reciente | ||
| Dist | SD | Dist | SD | |
| B. atratus | 0.5205 | 0.0018 | 4.059 | 0.006742965 |
| B. funebris | 1.2091 | 0.0028 | 1.133 | 0.002644156 |
| B. hortulanus | 0.2288 | 0.0013 | 0.248 | 0.00098899 |
| B. robustus | 0.7599 | 0.0019 | 0.632 | 0.001889028 |
| B. rubicundus | 0.5906 | 0.0014 | 0.813 | 0.002471686 |
Dist: Distancia, SD: desviación standard.
En la figura 2 se presentan las relaciones filogenéticas y relaciones entre haplotipos de las especies B. atratus, B. funebris, B. hortulanus, B. robustus y B. rubicundus obtenidas a partir del alineamiento 1, que incluyó 31 secuencias de 658 pb obtenidas a partir de material recientemente recolectado, y 23 secuencias ensambladas a partir de mini-barcodes de longitudes entre los 98 y los 658 pb. Se observa que todos los haplotipos generados a partir de especímenes de museo agrupan correctamente con las especies morfológicas a las que corresponden, formando grupos monofiléticos.
Por otro lado, dos grupos con buen soporte nodal, uno conformado por las especies B. funebris, B. hortulanus, B. robustus y B. rubicundus y otro conformado por los especímenes B. atratus evidencian la separación entre especies de cara corta y cara larga definida por en la longitud del área malar, previamente establecida por Williams, Cameron, Hines, Cederberg y Rasmont (2008) (Fig. 2A, Fig. 2B). Al interior del clado de la especie B. atratus se observan dos subclados bien diferenciados, uno compuesto por los haplotipos ATRA1 y ATRA7 (Fig. 2C) provenientes de muestras de museo, y el otro compuesto por los haplotipos ATRA 2, 3, 4, 5, 6 y 8 (Fig. 2D). En el clado D, los haplotipos 3, 4, 5 y 6 corresponden a secuencias de especímenes recientemente recolectados (frescos); mientras que los haplotipos ATRA2 y ATRA8, incluyen secuencias provenientes de especímenes de museo (Tabla 1). Esta diferenciación se ve reflejada en la red de haplotipos en la que el grupo conformado por los haplotipos ATRA1 y ATRA7 aislados de especímenes recolectados en el departamento de Antioquia, se encuentra separado por 16 pasos mutacionales del grupo conformado por los haplotipos restantes recolectados en otras localidades. Un caso similar se observa con la especie B. funebris, en la que, a pesar de la baja resolución de la filogenia, se evidencia que de la politomía que compone este grupo se deriva un subclado (E), conformado por los haplotipos FUN4, FUN5 y FUN6, correspondientes a especímenes recolectados en el departamento de Risaralda, en el PNN Los Nevados (Tabla 1). La red de haplotipos indica que entre los haplotipos arriba mencionados y los haplotipos FUN1, 2, 3, 7 y 8 pertenecientes a individuos recolectados en el PNN Chingaza, hay cuatro pasos mutacionales de distancia.
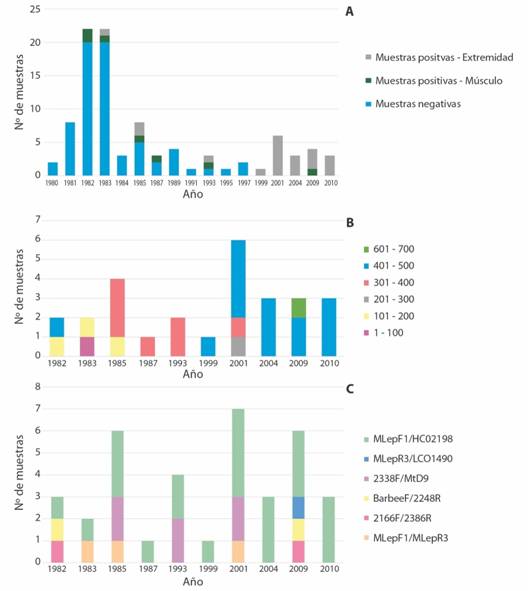
Fig. 2 Fig. 2 Relaciones filogenéticas del gen COI y redes de haplotipos de las especies Bombus atratus, B. funebris, B. hortulanus, B. robustus y B. rubicundus construidas a partir de un alineamiento de 658 pb. Los tonos claros indican secuencias provenientes de material recientemente recolectado, mientras que los tonos oscuros denotan secuencias obtenidas a partir de especímenes de museo. Sobre las ramas se presenta el soporte de cada clado (probabilidades posteriores), soportes inferiores a 0.80 no incluidos. Phylogenetic relationships of the COI gene and haplotype networks of the species Bombusatratus, B. funebris, B. hortulanus, B. robustus and B. rubicundus constructed from an alignment of 658 bp. Light tones indicate sequences from recently collected material, while dark tones denote sequences obtained from museum specimens. Above the branches, the support of each clade is presented (posterior probabilities), support values less than 0.80 not included.
Con respecto a los análisis realizados con fragmentos de diferentes tamaños amplificados a partir de las combinaciones de primers detalladas en la Tabla 2, se observó que al igual que en la reconstrucción filogenética realizada con el fragmento completo de 658 pb, todas las especies fueron reconstruidas como grupos monofiléticos, los cuales se encuentran separados de acuerdo con la característica morfológica en la longitud del área malar (cara larga o cara corta) (Apéndice Digital 3A, Apéndice Digital 3B). De este modo, aunque la topología general de todos los análisis es similar, solo en cuatro de los seis análisis, obtenidos con fragmentos correspondientes a los primers MLepF1/HCO2198, 2166F/2386R, 2338F/MtD9, MLepF1/MLepR3 (Apéndice Digital 3B, Apéndice Digital 3C, Apéndice Digital 3E y Apéndice Digital 3F) se reconstruyen los dos grupos observados para B. atratus y B. funebris en la topología obtenida con el fragmento completo de 658 pb. En el análisis realizado con los primers MLepR3/LCO1490 los haplotipos pertenecientes a la especie B. atratus se presentan como una politomía, mientras que la topología obtenida de secuencias amplificadas con los oligonucleótidos BarbeeF/2248R (Apéndice Digital 3A), el haplotipo MK544991 (ATRA1) se presenta separado del clado al interior del cual el haplotipo MK544992 (ATRA1) se encuentra incluido. A diferencia de lo anteriormente expuesto, las secuencias ATRA1 se presentan como grupo hermano del haplotipo ATRA7, formando los tres un clado separado de los demás haplotipos en la topología obtenida con el fragmento de COI completo (Fig. 2A). Adicionalmente es de resaltar que para el caso de B. funebris, los haplotipos son agrupados de acuerdo con el lugar en el que el espécimen fue recolectado. Por otro lado, empleando los oligonucleótidos MLepF1/MLepR3 (Apéndice Digital 3F), las especies B. hortulanus y B. robustus se presentan como parte de una politomía en la que a partir del nodo diverge un haplotipo de B. hortulanus, y dos clados, uno en el que se agrupan los haplotipos de B. robustus, y otro en el que se agrupan las secuencias restantes de B. hortulanus. Aunque los soportes nodales obtenidos en las diferentes topologías obtenidas son buenos, ninguno de los análisis realizados a partir de los diferentes mini-barcodes evaluados refleja la topología obtenida con el fragmento de 658 pb. No obstante lo anterior, la topología más cercana fue la obtenida con los primers MLepF1/HC02198.
Discusión
La combinación de información complementaria proveniente de diferentes disciplinas ha dado origen a lo que se denomina taxonomía integrativa (Dayrat, 2005), en respuesta a la necesidad de caracterizar a una escala más detallada la biodiversidad. Además de contribuir en la delimitación de especies, la taxonomía integrativa permite diseñar planes de conservación, así como políticas de uso sustentable de las especies (Lecocq et al., 2016a; Lecocq, Rasmont, Harpke, & Schweiger, 2016b), a partir del uso del conocimiento multidimensional de las especies.
Para el caso particular de Bombus, dada la habilidad de estas especies para la polinización de cultivos estos insectos han sido objeto de translocación con fines comerciales (Lecocq et al., 2016b); lo que ha generado un gran impacto sobre la fauna nativa de abejorros en diferentes países (Morales, 2007; Kanbe, Okada, Yoneda, Goka, & Tsuchida, 2008; Kraus et al., 2011; Morales, Arbetman, Cameron, & Aizen, 2013). Recientes estudios han puesto de manifiesto la existencia de una gran diversidad genética subyacente a las especies nominales de este género (ej. Williams et al., 2012; Françoso et al., 2019; Martinet et al., 2019). Sin embargo, para las especies de Bombus distribuidas en Colombia, hasta el momento la información disponible en bases de datos públicas como GenBank y BOLD es casi inexistente, por lo que una de las grandes contribuciones de este estudio es la generación de información genética del marcador mitocondrial COI por primera vez para las especies B. atratus, B. hortulanus, y B. rubicundus, y los primeros registros de este marcador para Colombia de las especies B. funebris, y B. robustus. Además, este es el primer estudio en Colombia, en el que se realiza la caracterización genética de especies de abejas nativas empleando especímenes preservados en una colección biológica en el que la mayoría de especímenes superan los 10 años de antigüedad; lo que según el criterio propuesto por Hernández-Triana et al. (2014), las hace poco adecuadas para estudios genéticos dada la degradación del material genético.
Por otro lado, a pesar del bajo número de individuos analizado, a partir del análisis de las secuencias obtenidas, se determinó gran diversidad genética intraespecífica al menos para dos de las cinco especies analizadas (Fig. 2, Tabla 3), lo que puede indicar la presencia de diferentes unidades de manejo (Moritz, 1994) bajo el nivel taxonómico de especie.
Calidad de ADN obtenido yéxito de amplificación: En el caso particular de este estudio, de 96 especímenes escogidos, se obtuvo ADN de cerca del 35 % de los individuos de los que se obtuvieron 23 secuencias que pudieron ser empleadas en los análisis de reconstrucción filogenética y de redes de haplotipos, generando hipótesis congruentes con la información obtenida de material recientemente recolectado. Se evaluaron 39 individuos recolectados y preservados entre 1980 y 1985 (de 30 a 31 años de edad al momento de la extracción), de los cuales se pudieron generar secuencias de 300 a 406 pb a partir del ensamblaje de diferentes fragmentos. En general el éxito de amplificación en muestras previas a 1985 fue bajo (7.6 %), lo que coincide con diferentes estudios en los que se concluye que uno de los factores que más influye en el éxito de amplificación de ejemplares de museo es el tiempo transcurrido a partir del depósito del espécimen en dichos acervos; así, la probabilidad de obtener material genético es inversamente proporcional a dicho tiempo (Hajibabaei et al., 2006; Gilbert et al., 2007; Hernández-Triana et al., 2014; Mitchell, 2015). No obstante, de los 57 individuos restantes, recolectados desde 1987 hasta el 2010, se obtuvo ADN y productos amplificables que pueden ser empleados como mini-barcode para todas las muestras, lo cual muestra cómo las colecciones biológicas constituyen un pilar importante para la taxonomía integrativa siendo repositorio de información de caracteres morfológicos, distribución previa y actual de especies e información genética que se encuentra contenida en cada uno de los especímenes preservados (Wandeler et al., 2007).
Nuestros resultados indican un mayor éxito y amplificación en muestras de extremidad derecha del mesotórax, en comparación con las muestras de músculo torácico procesadas (Fig. 1C). Adicionalmente, se observó una mayor integridad del ADN obtenido a partir de extremidad, lo que fue evidenciado en la obtención de fragmentos de entre 300 y 500 pb. Dicha integridad fue menor en muestras provenientes de individuos recolectados previo a 1983 (Fig. 1A). Reportes previos han indicado la reducción en el éxito en la extracción de ADN y subsecuente amplificación en especímenes montados en alfiler en la medida en la que la edad del espécimen (entendida como el tiempo que ha permanecido en la colección biológica tras su recolecta) aumenta; sin embargo, el tiempo en la de la viabilidad de estos especímenes es variable (Watts, Thompson, Allen, & Kemp, 2007; Strange, Knoblett, & Griswold, 2009).
Por otro lado, es de resaltar que los oligonucleótidos con los que se reportó un mayor número de amplificaciones, aunque no siempre se obtenía la totalidad del fragmento, fueron los primers MLepF1/HCO2198 (Fig. 1B). Dichos oligonucleótidos amplifican un fragmento de 407 pb de bases en el sector más variable de la secuencia hacia el extremo 3’, por lo que resultan de gran potencial en la identificación molecular de nuevas especies (Júnior et al., 2015).
Relaciones filogenéticas y divergencia genética: la totalidad de los especímenes estudiadosfue agrupada correctamente de acuerdo con la especie morfológica que les había sido asignada. No obstante, a partir de la incorporación de la información genética suministradas por los individuos de museo procesados, se pudo observar un incremento en la distancia genética inter-específica, particularmente con respecto a las especies B. atratus y B. funebris (Tabla 3). Esto se debe a la presencia de sustituciones nucleotídicas diferenciales a lo largo de las secuencias. En estudios recientes se ha reportado la incorporación de errónea de nucleótidos en cerca del 17 al 21 % de las secuencias amplificadas a partir de material de museo (Stiller et al., 2006; Sefc, Payne, & Sorenson, 2007; Wandeler et al., 2007), en el que las transiciones representan cerca del 60 % mientras que el nivel de transversiones es menor (Stiller et al., 2006). No obstante, para el caso particular de este estudio, pensamos que las sustituciones no son un artefacto de la antigüedad de las muestras, ya que estas fueron similares entre grupos de individuos, lo que se vio reflejado en la formación de dos subclados en las especies B. atratus y B. funebris (Fig. 2). Los linajes fueron agrupados en dichos subclados de acuerdo al lugar de proveniencia de las muestras. De este modo, para el caso de B. funebris, el clado compuesto por los haplotipos FUN4, FUN5 y FUN6 corresponde a individuos recolectados en la cordillera central colombiana mientras que las secuencias restantes corresponden a individuos recolectados en la cordillera oriental. Esto indica la presencia de al menos dos poblaciones que pueden constituirse en Unidades Significativas Evolutivas según Moritz (1994). El aislamiento entre estas poblaciones puede deberse al rango de distribución altitudinal restringido de la especie (2 850-4 750 m.s.n.m., Lievano et al., 1991), que posiblemente limitó su desplazamiento hacia las tierras bajas durante los cambios ambientales ocurridos en el Pleistoceno, y que causó aislamiento reproductivo de las poblaciones provenientes de dos cordilleras diferentes. En contraste, las poblaciones de la especie B. atratus, no son frecuentes en los pisos montanos altos, lo que eventualmente limita el contacto entre las poblaciones provenientes de los dos lados de la cordillera. Así al interior de esta especie también se pudo determinar la presencia de linajes mitocondriales únicos, como es el caso del grupo compuesto por los haplotipos ATRA1 y ATRA7 recolectados en el departamento de Antioquia, que pueden ser considerados como unidades evolutivas significativas (ESU), con prioridad de conservación (Moritz, 1994). Nuestros resultados coinciden con estudios previos realizados en Brasil con la especie B. pauloensis (sinónimo taxonómico de B. atratus), en los que se determinó la presencia de diferentes grupos genéticos al interior de esta especie, que pueden ser consideradas como unidades de manejo, para las cuales se ha previsto que presenten diferentes comportamientos bajo un escenario de cambio climático (Françoso et al., 2019).
Sugerencias para el protocolo de extracción de DNA de insectos depositados en colecciones biológicas (museos): Se determinó que el almacenamiento de los especímenes una vez sacrificados influye en gran medida en la preservación del material genético contenido en ellos. De este modo, se registró que los individuos almacenados en ambientes húmedos son más proclives al daño del ADN por la actividad de las endonucleasas, que se mantienen activas durante largo tiempo después de la muerte del espécimen (Dessauer, Cole, & Hafner, 1990). Por tal motivo, se recomienda secar los especímenes para inactivar las endonucleasas y así inhibir la degradación (Doyle & Dickson, 1987), pero esta desecación también tiene desventajas a la hora de emplear el material genético del espécimen, pues tras la desecación, el ADN en los tejidos blandos pasa por un proceso de oxidación, que particularmente en el caso de las pirimidinas causa la fragmentación del anillo, lo que ha sido evidenciado en perfiles cromatográficos de ADN realizados en muestras secas de tejido almacenadas durante largos periodos de tiempo (Pääbo, 1989). Esto podría explicar por qué la extracción de ADN fue más exitosa de extremidades, pues para cortar la extremidad de los especímenes, estos no tuvieron que ser sometidos a cámara húmeda, mientras que para la extracción de músculo fue necesario humedecerlos, lo que pudo dañar aún más el ADN. De este modo se puede inferir también que próximas extracciones de tejido de estos individuos pueden presentar resultados aún menos eficientes.
Una alternativa a la desecación puede ser preservar la muestra embebida en fluidos diferentes a la formalina o Carnoy, ya que estas sustancias son conocidas por causar daño en el ADN (Dillon, Austin, & Bartowsky, 1996; Wandeler et al., 2007). Particularmente en insectos se usa el almacenamiento en alcohol, de hecho, Dillon et al., (1996) extrajeron ADN de buena calidad de muestras almacenadas a 24 meses a 4 °C en etanol 70 %. Del mismo modo obtuvieron ADN en calidades y cantidades similares a las muestras frescas cuando los individuos fueron almacenados en alcohol absoluto. Así pues, se recomienda para posteriores iniciativas de aprovechamiento del material de la colección biológica, que una vez sean recolectadas las muestras, una parte del tejido o un individuo completo pueda ser almacenado en etanol absoluto, y que el medio sea reemplazado periódicamente para evitar el daño del ADN dentro de la muestra. Adicionalmente, aunque en este estudio no se evaluó el impacto de las sustancias empleadas en el proceso de eutanasia de los especímenes; Dillon et al. (1996), indican que el sacrificio practicado con Etil Acetato causa daños en el ADN que impiden la amplificación en especímenes recientes, mientras que Dean y Ballard (2001) lograron la extracción de ADN a partir de especímenes recientemente sacrificados de Drosophila simulans. Adicionalmente, estos autores concluyen que, aunque el método de sacrificio afecta la extracción de ADN, no afecta la amplificación.
En cuanto a la amplificación, además de la incorporación errónea de nucleótidos en las secuencias al momento de la amplificación (Stiller et al., 2006; Sefc et al., 2007; Wandeler et al., 2007), también se ha registrado la formación de productos quiméricos a través de un fenómeno denominado “jumping PCR” en las amplificaciones realizadas con material proveniente de colecciones biológicas (Pääbo, Irwin, & Wilson, 1990; DeSalle, Barcia, & Wray, 1993). Estos productos pueden derivarse de contaminación cruzada con ADN exógeno (Joly, Stevens, & van Vuuren, 2007; Wandeler et al., 2007) o a polimorfismos, o si por el contrario está relacionada con la formación de productos quiméricos, es altamente recomendable seguir las recomendaciones de los protocolos de trabajo sugeridas por Wandeler (et al., 2007), con el objeto de evitar la contaminación cruzada. Dichas modificaciones consisten en realizar la preparación de las reacciones en espacios que fueron previamente esterilizados (ej., con luz U.V.), así como emplear reactivos y consumibles exclusivamente para trabajo con ADN, y evitar el procesamiento de grandes cantidades de muestra. Adicionalmente, se recomienda realizar varias amplificaciones independientes, hacer uso de enzimas de alta fidelidad o técnicas como la clonación con el fin de detectar y evitar la incorporación de nucleótidos de manera errónea, y/o productos quiméricos en las secuencias empleadas para inferencia filogenética y otros estudios.
En Colombia los estudios orientados hacia la caracterización genética de las especies de Bombus son escasos, no existe un indicador directo del grado de conservación de los abejorros en el país o el impacto que el incremento de la urbanización, uso de pesticidas, o la fragmentación de hábitat entre otras prácticas ha causado sobre este género (Ospina & Nates, 2016). Es en este punto en el que la colección biológica del Laboratorio de Investigaciones en abejas de la Universidad Nacional de Colombia-LABUN, así como las diferentes colecciones entomológicas en el país revisten gran importancia como reservorio de información sobre la distribución histórica de las especies allí catalogadas. Para el caso particular de este estudio muestras de museo sirvieron como fuente de información genética de cada una de las poblaciones estudiadas. De este modo, en el presente estudio además de generar un protocolo de extracción y amplificación de secuencias código de barras a partir de especímenes depositados en la colección hace más de 30 años, se determinó en una pequeña muestra la presencia de haplotipos únicos al interior de dos especies nativas B. atratus, B. funebris, los cuales pueden constituirse en unidades evolutivas significativas diferentes sujetos de conservación (Moritz, 1994) o incluso en subespecies (caso de B. atratus). Estudios usando marcadores moleculares en otros lugares del mundo han demostrado la existencia de un amplio espectro de variaciones genéticas al interior de las especies del género Bombus, que a su vez están asociadas con una distribución geográfica determinada y que posiblemente presentan adaptaciones propias para la explotación de los entornos en los que viven. (ej. Williams et al., 2012; Françoso et al., 2019). Estudios como el presente resaltan la necesidad de explorar de una manera más profunda la diversidad en todos sus componentes (número de linajes evolutivos, diversidad genética y funcional (Pérez & García, 2001), pues a partir de la integración de esta información, se hace posible el planteamiento de medidas para la conservación de las poblaciones de estos polinizadores frente a escenarios de cambio climático, o iniciativas de explotación comercial y translocación de especies (Lecocq et al., 2016a; Françoso et al., 2019; Lotta-Arevalo, 2019, en prensa).
Declaración de ética: los autores declaran que todos están de acuerdo con esta publicación y que han hecho aportes que justifican su autoría; que no hay conflicto de interés de ningún tipo; y que han cumplido con todos los requisitos y procedimientos éticos y legales pertinentes. Todas las fuentes de financiamiento se detallan plena y claramente en la sección de agradecimientos. El respectivo documento legal firmado se encuentra en los archivos de la revista.
Apéndice digital 1
