










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.56 n.3 San José Sep. 2008
Hemoglobinas anormales en la población neonatal de Costa Rica
Gabriela Abarca1, Marta Navarrete3, Rafael Trejos1, Carlos de Céspedes1,2 & Manuel Saborío1,2
1. Programa Tamizaje Neonatal. Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera", A.P. 7786-1000, San José, Costa Rica. Fax (506) 225-825-16; gatamizaje@yahoo.es
2. Servicio de Genética Médica y Metabolismo, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera", San José, Costa Rica.
3. Servicio de Oncohematología, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera", San José, Costa Rica.
Abstract: Abnormal haemoglobins in the newborn human population of Costa Rica. Hemoglobinopathies are hereditary autosomic recessive diseases. A total of 70 943 samples of whole blood collected by heel prick in filter paper (S&S 903) from throughout Costa Rica (October 2005-October 2006) were analyzed to detect variants of hemoglobin by the iso-electric focusing technique. Eight hundred ninety one cases presented some variant, for a frecuency of 1/79. Five cases are homozygous for hemoglobin S (sickle cell disease) and one shows the double heterozygous genotype SC. in this study the S and C variants of hemoglobin, although with some local differences, are widespread all over the country. Thus, the prevention of new cases is important through the testing of hemoglobin in the Costa Rican National Newborn Screening Program, together with a interdisciplinary National Program of Education for the disease and carrier status (AS/AC) for patients, families and medicar personnel. This is the basis for proper genetic counseling, to improve treatment and to reduce morbi-mortality. Rev. Biol. Trop. 56 (3): 995-1001. Epub 2008 September 30.
Key words: hemoglobinopathy, neonatal screening, sickle cell disease, iso-electric focusing technique, hemoglobin S, hemoglobin C, Costa Rica.
Las hemoglobinopatías son trastornos hereditarios autosómicos recesivos del gen de la hemoglobina. Se debe distinguir entre el portador heterocigoto asintomático que solamente presenta de la afección de un alelo del gen de la globina pero con capacidad de transmitir el defecto a sus descendientes y los homocigotos o dobles heterocigotos que sí sufren la enfermedad (Davies et al. 2000).
Las hemoglobinopatías son producto de un trastorno de la molécula de hemoglobina (Hb) que pueden ocurrir por el simple cambio de un aminoácido en una de las cadenas (alfa o beta) de la globina, o por la falta de síntesis (total o parcial), de una de estas cadenas. En el primer caso se designan como hemoglobinopatías estructurales por mutación puntual del ADN, en el segundo caso se denotan como talasemias. Ambas alteraciones cualitativas y cuantitativas pueden coexistir produciendo las hemoglobi nopatías talasémicas -HbS+beta Tal- (Martínez et al. 1997).
Los defectos en la molécula de hemoglobina afectan millones de personas en todo el mundo, causando un alto grado de morbimortalidad (Malcorra 2001). En la actualidad se han descrito 600 variantes (Davies et al. 2000), y se considera como el error congénito más frecuente en poblaciones africanas, del mediterráneo, Asia, Caribe, América Central y del Sur.
Se ha calculado para América Latina una probabilidad de anemia falciforme (HbSS) de 0.16 /1000 y para talasemia de 0.1/1000. En poblaciones de origen africano-caribeño se ha calculado una frecuencia de 6.1% a 18% para HbS y de 0 a 5% para HbC (OPS 1987, Martínez et al. 1997, Dulin et al. 2003, OMS 2005).
No todas las hemoglobinopatías producen manifestaciones clínicas, sin embargo cuando éstas ocurren son variadas, presentándose en forma asintomática, leve o severa, como es el caso de la Hemoglobinopatía S homocigota, que presenta cuadros de anemia hemolítica crónica.
Estudios poblacionales multirraciales dan lugar al conocimiento de nuevas variantes de hemoglobina no descritas anteriormente así como nuevas distribuciones geográficas probablemente por el movimiento poblacional a lo largo del mundo.
Actualmente se acepta que América estaba libre de malaria y exenta de hemoglobinopatías antes de la llegada de los europeos. Esto fue demostrado en estudios de amerindios de América del sur que viven en zonas infestadas de Plasmodium y presentaron homocigosis para hemoglobina A (AA) (Sáenz 2005).
El flujo génico de la HbS en América proviene exclusivamente del África, a excepción de una pequeña fracción aportada por inmigrantes europeos. Los genes talasémicos tienen un origen más amplio, fueron introducidos por europeos y en menor proporción por africanos y asiáticos (Martínez et al.1985.)
En Costa Rica la búsqueda intencional de hemoglobinas anormales se inicia desde 1966 (Sáenz 2005) al realizar un estudio en la población del cantón de Santa Cruz, Guanacaste detectando un 7.5% de muestras positivas por drepanocitosis y un caso homocigoto para hemoglobina S.
El análisis e investigación de las hemoglobinas en nuestro país comienza en los años 80 con la creación del Centro de investigación en Hemoglobinas Anormales y Trastornos Afines (CIHATA) de la Facultad de Microbiología de la Universidad de Costa Rica, gracias a ello se han descubierto varias hemoglobinas autóctonas ( Sáenz 2005 ).
En la Actualidad el CIHATA realiza técnicas en biología molecular (detección de halotipos) por lo que es posible rastrear la procedencia de las mutaciones en la hemoglobina, proporcionando información antropológica que relaciona el cuadro clínico con la sobrevida del paciente (Rodríguez et al. 1998).
En octubre 2005, el Programa Nacional de Tamizaje Neonatal inicia pioneramente la detección de hemoglobinas anormales en toda la población neonatal costarricense, con una cobertura actual del 99%, con el fin de detectar y tratar a los enfermos por hemoglobinas anormales desde el nacimiento, favoreciendo su tratamiento y seguimiento con claras ventajas para pacientes y sus familiares.
El presente trabajo tiene el objetivo de establecer la incidencia de las diferentes hemoglobinopatías en la población neonatal nacida en Costa Rica desde Octubre 2005 a Octubre 2006 como un aporte para conocer mejor la realidad nacional de las hemoglobinopatías, destacando la importancia de su detección en la población neonatal.
Materiales y métodos
Población estudiada: Se analizó un total de 70 943 muestras de sangre total en papel filtro S&S 903, proveniente de neonatos de Costa Rica. Las muestras ingresaron al Laboratorio de Tamizaje Neonatal de Costa Rica entre octubre 2005 y octubre 2006.
Obtención del espécimen: La detección de hemoglobinas anormales se realiza utilizando sangre obtenida por punción del talón en los 4 a 7 días posteriores al nacimiento del niño(a). Las muestras provienen principalmente de cada uno de los 718 centros de recolección ubicados en todo el país (Hospitales, Clínicas, Áreas de Salud y EBAIS) gracias a la Seguridad Social (CCSS).
La detección de variantes de hemoglobina se lleva a cabo mediante la técnica de isoelectroenfoque que separa las diferentes proteínas de la hemoglobina en bandas, a través de su punto isoeléctrico (carga neta de la proteína es cero). Toda muestra que presente una banda sospechosa es reanalizada por el mismo sistema de isoelectroenfoque. Si la positividad permanece se recolecta muestra del padre, la madre y el neonato para posterior análisis por punto isoeléctrico. Los casos homocigotos o doble heterocigoto son referidos al Servicio de Hematología del Hospital Nacional de Niños para tratamiento y seguimiento. Los casos de portadores heterocigotos son notificados al centro de recolección de donde procede la muestra para consejo genético a la familia.
Procedimiento de laboratorio: Las muestras fueron tratadas con 18 µL del reactivo de elusión del kit para hemoglobinas de la casa Perkin Elmer por espacio de 30 minutos.
Se procede a realizar una electroforesis de agarosa que contiene anfolitos que permiten un gradiente de pH 6-8 favoreciendo la distribución de los diferentes puntos isoeléctricos, bajo las siguientes condiciones: 1400 V, 300mA, 40 watts, por espacio de una hora treinta minutos, posterior a esto, se precipita la proteína del gel con ácido tricloroacético al 10 % (10 minutos) para luego lavarlo con agua destilada por una hora con cambios del líquido cada 15 minutos.
Se realiza el secado del gel en un secador de geles FR-9610 Perkin Elmer por espacio de una hora y se obtienen los resultados confirmados, al repetir muestras sospechosas por duplicado.
La cuantificación de la hemoglobina se realiza con el sistema Perkin Elmer isoScan imagen.
Para asegurar la garantía de calidad se mantiene un control externo con los Centers for Disease Control (CDC) en Atlanta (EEUU), el informe del resultado incluye el fenotipo y el diagnóstico clínico de cada una de las cinco muestras recibidas trimestralmente.
Resultados
De las 70 943 muestras de sangre seca de neonatos se detectaron 897 casos con alguna variante de hemoglobina para una frecuencia de 1/79 .Es importante destacar que la cobertura del estudio comprende el 99 % de neonatos nacidos en el país por lo que los valores obtenidos pueden extrapolarse a cifras muy cercanas a la incidencia real.
Geográficamente la provincia que presenta mayor porcentaje de casos es San José (23.3%), seguido por las provincias de Guanacaste (18.1%) y Limón (17.2%). En menor proporción Alajuela y Puntarenas presentan valores de 14% y 11.8% respectivamente. Heredia y Cartago son la provincias con menor afección con un 5.8% y 4.8%, respectivamente (Cuadro 1).
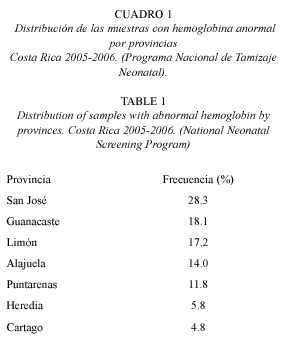
De acuerdo al tipo de hemoglobina anormal encontrada en nuestra población; se clasificó 5 casos como homocigotos para hemoglobina S con fenotipo neonatal (FSS) que corresponde a una frecuencia de 1/14 (189 tamizados). En el caso del fenotipo neonatal FCC no se detectó ninguno, y para el caso de doble heterocigoto (FSC) se detectó uno solo con una frecuencia de 1/70 943.
El rasgo falciforme fenotipo neonatal FAS se encontró en 765 casos, para hemoglobina C en 53, para hemoglobina D en 25 y en 3 casos para hemoglobinas indeterminadas. Las frecuencias fueron respectivamente: FAS 1/93, FAC 1/1339, FAD 1/2838 y FAX 1/23648 (Cuadro 1).
Los falsos positivos tuvieron una frecuencia de aparición de 1/1 819.
Todos los niños homocigotos y doble heterocigoto fueron remitidos al Servicio de Hematología del Hospital Nacional de Niños para ser incluidos en el protocolo de tratamiento y seguimiento de estas enfermedades.
Se encontró un 15 % de muestras (109 casos) de niños con padres de procedencia extranjera.
La distribución por provincia para hemoglobina S fue la siguiente: de los 765 casos detectados la mayor proporción corresponde a San José con 209 (27.3%) seguido de Guanacaste con 144 casos (18.8%) y Limón con 126casos (16.5%). Alajuela y Puntarenas lo presentó en menor grado 116 casos (15.2%) y 92 casos (12%). Heredia y Cartago son las provincias menos afectadas 45 casos (5.9%) y 33 casos (4.3%) respectivamente.
En el caso de la hemoglobina C se detectaron 53 portadores y se encontró una mayor proporción en Limón 18 casos (34.1%), seguido de San José 17 casos (32%), Guanacaste 5 casos (9.4%), Alajuela y Puntarenas con 4 casos cada una (7.5%), Heredia con 3 casos (5.7%) y Cartago con 2 casos (3.8%).
La hemoglobina D fue detectada en todas las provincias con la siguiente distribución: 9 casos para San José (36%), 6 casos en Guanacaste (24%), seguido de 3 casos en Puntarenas (12%) y Cartago. En Alajuela 2 casos (8%) y Limón y Heredia solamente 1 (4%) (Cuadro 2).

Discusión
En el continente americano, la presencia de hemoglobinas anormales es producto de la inmigración y entrecruzamiento de diversas poblaciones europeas y africanas con los grupos amerindios residentes, provocando una mezcla racial con la introducción de mutaciones en el gen de la hemoglobina. La accesibilidad de las comunicaciones junto con el tránsito humano ha favorecido que la población blanca americana (Martínez et al. 1985, Sáenz 2005) presente dichas alteraciones por lo que la presencia de hemoglobina S o de otras hemoglobinas anormales no debe relacionarse únicamente con el grupo étnico negro (Rodríguez et al. 1998).
La frecuencia de hemoglobina S en América tropical es de 71.8% (Malcorra 2001, Machín et al.2004).
En este estudio los datos demuestran que la población neonatal costarricense exhibe dos importantes mutaciones en su molécula de hemoglobina presentando características fenotípicas de hemoglobina S y hemoglobina C en mayor proporción y unos pocos casos para hemoglobina D.
Hemoglobina S, ampliamente distribuida por todo el país, se exhibe con mayor frecuencia en la población de San José, resultado probablemente de la migración de individuos de las provincias hacia el centro de la capital al igual que la permanencia de extranjeros (nicaragüenses, panameños, caribeños españoles, orientales, entre otros) quienes se mezclan con citadinos generando nuevas mezclas génicas de las variantes ya existentes en la zona.
Guanacaste es la segunda provincia que exhibe mayor cantidad de casos de hemoglobina S. El principal reservorio de la mutación para hemoglobina S se encuentra en África Central y ésta llega a nuestro país por .medio del trasiego de grupos étnicos negros de Cuba hacia nuestro país y al influjo migratorio de negros procedentes de Panamá y Nicaragua, así como esclavos que acompañaron a los conquistadores en los albores de la colonia. Heredia y Cartago no están exentos de hemoglobina S pero son ciudades menos influidas por migraciones de esclavos negros en épocas de la colonia (Rodríguez et al. 1998).
Limón ocupa el tercer lugar de frecuencia en casos para hemoglobina S, probablemente por la misma migración de sus pobladores hacia la zona central del país u otras zonas lo que favorece la amplia distribución de esta mutación.
Estos datos indican que el tamizaje de hemoglobinas anormales debe orientarse y realizarse en toda la población tanto caucásica como étnia negra por que ambas están en riesgo.
Para la mutación de la característica fenotípica C, Limón presenta la mayoría de casos, en segundo lugar San José y en tercer lugar Guanacaste.
La mutación en la molécula de hemoglobina para la característica C es originaria de África Occidental. En Costa Rica una parte importante de la población negra del Atlántico provenía de zonas de Benín, y territorios circundantes africanos con alta frecuencia de hemoglobina C (Sáenz 2005). La zona de Limón por ser portuaria recibe mucha influencia extranjera lo que favorece la aparición de esta característica fenotípica.
En las islas y regiones que forman la Cuenca del Caribe la frecuencia de HbC es de 1.2 a 6.4%. (Malcorra 2001, Machín et al. 2004, Salazar 2004).
La característica fenotípica S es mucho más frecuente en el país que la C, sin embargo la hemoglobina C también se encuentra distribuida por todo el país.
La característica fenotípica D, originaria de la india se detectó en solo 25 casos.
De los casos detectados como enfermos observamos 5 del fenotipo FSS y solamente uno del fenotipo FSC.
El curso clínico de la enfermedad por hemoglobina S se caracteriza por una anemia crónica con episodios intercalados de crisis hemolítica. En ausencia de las crisis la sintomatología anémica es escasa. La gravedad del cuadro clínico depende en parte de la concentración de hemoglobina fetal ya que cuanto mayor sea ésta, menor será la posibilidad de que el eritrocito experimente alteraciones irreversibles de su forma y función. La mayoría de los pacientes sufren trastornos constitucionales con retraso en el crecimiento y las manifestaciones clínicas son consecuencia de las crisis vaso-oclusivas producidas por la obstrucción del sistema vascular por los agregados de eritocitos.
Las crisis suelen desencadenarse por infecciones bacterianas o víricas, deshidratación, desoxigenación o frío y se acompañan de dolor abdominal, dolor articular, óseo y pleurítico. Los fenómenos oclusivos de la circulación cerebral y ósea son los más graves. Se pueden producir convulsiones, déficit neurológico grave e incluso coma. Oclusiones vasculares en los huesos favorecen la aparición de área de infarto sobre todo en vértebras y necrosis aséptica de la cabeza del fémur. Las manifestaciones viscerales afectan todos los órganos y sistemas. La insuficiencia sanguínea produce infartos en la micro circulación del ojo e infartos hepáticos. Las alteraciones circulatorias cutáneas favorecen la aparición de úlceras, con mayor frecuencia en los tobillos (Martínez et al. 1997).
Una de las complicaciones más graves la constituyen las crisis aplásticas que pueden deberse a infección por parvo virus o a un déficit de folatos.
El tratamiento se dirige a la prevención de las crisis, evitando las infecciones, la deshidratación, la estasis circulatoria y el frío. La oxigenoterapia no mejora el cuadro clínico. Fármacos que aumenten la síntesis de hemoglobina fetal como la hidroxiurea parecen tener buen resultado en el tratamiento de las drepanocitosis. Según lo señalado anteriormente, la administración de suplementos de ácido fólico es importante.
La Hemoglobinopatía SC produce un cuadro clínico menos grave que la HbSS, el crecimiento y desarrollo sexual son normales, la anemia es leve y las crisis convulsivas escasas, sin embargo, la afección retiniana es más grave que en HbSS y los accidentes trombóticos son más frecuentes.(Malcorra 2001).
La característica fenotípica de portador (rasgo) de una hemoglobina anormal (AS, AC) raras veces presenta sintomatología o alteraciones del hemograma a menos que las condiciones ambientales sean extremas, como hipoxia o deshidratación. El rasgo drepanocítico no requiere tratamiento (AS, AC).
La hemoglobina D como característica fenotípica de portador no presenta trastorno clínico alguno (estado heterocigoto AD), pero en estado homocigoto (DD) produce una anemia hemolítica discreta, y aunque no se producen alteraciones clínicas el hemograma es semejante al de las talasemias y se puede observar microcitosis discreta.
El porcentaje de mortalidad de niños menores de 5 años enfermos por drepanocitosis es del ámbito del 25 al 30% produciéndose secundariamente a infecciones fatales, secuestro esplénico o crisis aplásticas (Laird et al. 1996, Machín et al. 2004).
Este estudio demuestra una amplia distribución de portadores de variantes de hemoglobina que favorece la aparición de nuevos casos de enfermos sobre todo para la característica fenotípica S y C. Además nos lleva a pensar en la aparición de nuevas variantes genéticas producto de mezclas raciales que podrían favorecer estados de enfermedad. Con el tamizaje masivo neonatal para hemoglobinas en la población costarricense se hace factible la detección temprana de la anemia drepanocítica y otras hemoglobinopatías por defectos en cadenas alfa y beta, lo que permite tratarlas y controlarlas desde el inicio de su aparición, favoreciendo no solo la sobrevida del paciente si no significativamente una mejor calidad de vida. Por otro lado, la detección de portadores junto con un programa de educación sanitaria con el consejo genético prevendría la aparición de nuevos casos.
La vigilancia epidemiológica y la educación sanitaria deben actuar a nivel comunitario a través de los sistemas de atención primaria existente favoreciendo la concientización de la población con respecto al problema y prolongar la supervivencia de los afectados con mejor calidad de vida.
Unido a la detección temprana que realiza el Programa de Tamizaje Neonatal se requiere establecer un Programa Nacional Educativo interdisciplinario dirigido a médicos generales, enfermeros y pediatras de los diferentes centros hospitalarios, Clínicas y EBAIS de todas las zonas del país para orientarlos hacia un mejor manejo del paciente en el proceso de crisis y consejo genético a las familias portadoras o que padezcan la enfermedad.
La efectividad de estos programas establecidos en otros países como EEUU, Brasil España, inglaterra ha sido demostrada ( Pereira 1996, Davies et al. 2000, Dulín et al. 2003, Salazar 2004,).
El análisis de costo por prueba por individuo para programas de tamizaje de hemoglobinopatías que se realicen con una población neonatal sobre los 25 000 nacimientos por año hasta 40 000 nacimientos por año, y que utilizan técnicas de isoelectroenfoque y HPLC (Cromatografía líquida de alta resolución) son muy similares. El beneficio del HPLC radica en que se puede trabajar muestras a gran escala en programas de tamizaje neonatal, sin embargo, el isoelectroenfoque provee más información por tener una mayor resolución en la separción de bandas (Davies et al. 2000). Muchos países han optado por utilizar ambos métodos para mejorar el diagnóstico de laboratorio y orientar mejor la clínica.
Si nos enfocamos en el costo-beneficio para el paciente, el valor de la vida y las posibles secuelas que un niño con hemoglobinopatía haya padecido (como meningitis bacteriana con sordera, ceguera, retraso psicomotor entre otras) es incalculable por qué la detección temprana está más que justificada. . La detección precoz permite programas de seguimiento específicos y mejor manejo de los enfermos por parte del personal de salud y los familiares, que aseguran la reducción de la morbi-mortalidad.
Es importante comprender que el objetivo real costo beneficio debe ser visto en términos de beneficio del afectado y sus familiares.
Resumen
Se han analizado un total de 70 943 muestras de sangre total en papel filtro S&S 903 de neonatos de Costa Rica (octubre 2005 a Octubre 2006) con el fin de detectar variantes de hemoglobina mediante la técnica de isoelectroenfoque. Se detectaron 891 casos con alguna variante para una frecuencia de 1/79. Se clasifican 5 casos homocigotos para hemoglobina S (anemia drepanocítica o anemia falciforme) y un caso doble heterocigoto para SC. En este estudio se demuestra que las variantes fenotípicas de hemoglobina S como la C, se encuentran distribuidas por todo el país con algunas diferencias locales, razón por la cual es importante que la prevención de nuevos casos se realicé a través de nuestro Programa Nacional de Tamizaje de Hemoglobinas junto con un Programa Nacional interdisciplinario de Educación para el portador del rasgo (AS/AC) como, para el enfermo y su familia; al igual que la instauración de programas dirigidos a médicos generales y enfermeras en todas las regiones de salud del país, para asegurar consejo genético a portadores y enfermos, y a la vez, mejorar los sistemas de tratamiento a los pacientes para reducir la morbi -mortalidad.
Palabras clave: hemoglobinopatías, tamizaje neonatal, anemia drepanocítica, isoelectro-enfoque. Hemoglobina S, Hemoglobina C, Costa Rica.
Recibido 22-II-2007. Corregido 30-VI-2008. Aceptado 31-VII-2008.
Referencias
Davies, S., E. Cronin, M. Gill, P. Greengross, M. Hickman & C. Normand. 2000. Screening for sickle cell disease and thalassaemia: a systematic review with supplementary research. Health Technol Asses. 4: 55-66. [ Links ]
Dulin, E., M. Cantalejo, M. De Julián, & P. Galarón. 2003. Detección precoz neonatal de anemia falciforme y otras hemoglobinopatías en la comunidad autónoma de Madrid. Estudio piloto. Ann Pediatr. 58: 146-155. [ Links ]
Laird, L., C. Dezateux, E. Anionwu. 1996. Neonatal screening for sickle cell disorders: what about the carrier infants. Brit Med J. 313: 407-411. [ Links ]
Machín S., T. Guerra, E. Svarch, E. Espinoza, J. Mesa, E. Dorticós, A. Plasencia, A. González, A. Núñez & M. Wade. 2004. Morbiletalidad en pacientes adultos con drepanocitosis. Rev Cubana Hematol inmunol Hemoter. 20: 4-11 [ Links ]
Malcorra, J. 2001.Hemoglobinopatías y Talasemias. Can Ped. 25:265-277. [ Links ]
Martínez, G, A. Hernández, L. Corral, A. Muñiz, & A. Hernández. 1997. Biología molecular de la hemoglobinopatías y hemopatías malignas. Rev Cubana Hematol inmunol Hemoter. 12: 3-6. [ Links ]
Martínez, G., B. Colombo. 1985. Hemoglobinopatías en Cuba. Rev Cubana Hematol inmunol Hemoter. 1: 32-36. [ Links ]
OPS, OMS. 1987. Informe de la reunión de un grupo de expertos en genética médica Genética en América Latina y el Caribe. Ejecución de las actividades. La Habana, Cuba. 30p. [ Links ]
OMS. 2005. Prevalencia de la anemia falciforme. Reunión EB117/34. Génova, Francia. 195 p. [ Links ]
Pereira, F, I. Sáenz. 1996. Hemogobinopatías en niños. Colom. Médic. 27: 146-149. [ Links ]
Rodríguez, W., G. Sáenz, M. Chaves. 1998. Halotipos de la hemoglobina S: importancia epidemiológica, antropológica y clínica. Pan. Am. J. Public Health 3: 1-8. [ Links ]
Sáenz, G. 2005. Hemoglobinas anormales. Acta Méd Costarric. 47: 173-179. [ Links ]
Salazar, R. 2004 La Hemoglobina S en la población venezolana. Invest Clin. 45: 11-21. [ Links ]

