










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.52 n.3 San José Sep. 2004
Molecular diagnosis of hemophilia A and B. Report of five families from Costa Rica
Lizbeth Salazar-Sánchez1 , Guillermo Jiménez-Cruz2 , Pilar Chaverri2 , Winnie Schröder3 , Karin Wulff3 , Gerardo Jiménez-Arce1 , Miriam Sandoval2 , Patricia Ramírez2 & F.H. Herrmann3
1 Centro de Investigación en Hematología y Trastornos Afines (CIHATA), Facultad de Microbiología. Universidad de Costa Rica, San José, Costa Rica. Fax (506)223.1385; lsalazar@cariari.ucr.ac.cr
2 Servicio de Hematología, Hospital México, San José, Costa Rica.
3 Institute of Human Genetics; Ernst-Moritz-Arndt-University, Greifswald, Germany.
Received 01-VII-2003. Corrected 11-X-2003. Accepted 24-XI-2003.
Abstract
Hemophilia Aand B are X-chromosome linked bleeding disorders caused by deficiency of the respective coagulation factor VIII and IX. Affected individuals develop a variable phenotype of hemorrhage caused by a broad range of mutations within the Factor VIII or Factor IX gene. Here, were report the results of the molecular diagnosis in a five Costa Rican families affected with Hemophilia. Methods of indirect and direct molecular diagnosis are applied in three Hemophilia A and two Hemophilia B families from Costa Rica as well as preconditions, practicability and facilities of this diagnosis. In two families with Hemophilia A and both families with Hemophilia B the causative mutation could be detected by Southern blotting, polymerase chain reaction or sequence analysis. One Hemophilia A family could only analyzed by linkage analysis using genomic markers. Rev. Biol. Trop. 52(3): 521-530. Epub 2004 Dic 15.
Key words: Hemophilia A, Hemophilia B, factor IX, factor VIII, molecular diagnosis, carrier detection.
Palabras clave: Hemofilia A, Hemofilia B, factor IX, factor VIII, diagnóstico molecular, detección de portadores.
Hemophilia A and B due to factor VIII (FVIII) and IX (FIX) deficiencies are the most common bleeding disorders with incidences of about 1 per 4000 to 7000 and 1 per 25000 to 30000 male births respectively (Forbes et al. 1997). Since the diseases follow an X-linked recessive mode of inheritance, they mostly afflict men whereas most female carriers remain healthy. The genes for both coagulation factors are located at the distal end of the long arm of the human X-chromosome (Xq28 and Xq27) (Furie and Furie 1990). Theoretically carriers have a clotting factor level around 50% of normal, which is generally sufficient for normal haemostasis. So far, the application of clotting and immunological methods allow to detect only about 80% of the carriers due to the random inactivation of one X-chromosome in females. Anxiety about the risk of Hemophilia affecting their offspring is the reason, why possible carriers ask for counselling.
The precondition for an effective counselling is reliable detection of female carriers.
Molecular diagnosis of this disease as well as a carrier and prenatal diagnosis is possible since the FVIII and FIX gene where cloned and characterized (Furie and Furie 1990). The gene which codes for the FVIII protein is 186 kb long and has 26 exons which code for the FVIII protein (Gitschier et al. 1984). In molecular diagnosis of families with severe Hemophilia the natural starting point today is to ascertain whether the disease is due to inversion in the X-chromosome. Almost 45% of cases of severe Hemophilia A are caused by a substantial re-arrangements in the long arm of the X-chromosome. Two sequence elements homologous to intron 22 of the factor VIII gene are located in reverse orientation telomeric of the FVIII gene. Pairing and crossing over between the intron 22 element and the distal or proximal one of the extragenic elements result in an inversion of the FVIII gene with one break point in intron 22 (Fig. 1) (Pattinson et al. 1990, Lakich et al. 1993, Herrmann and Scharrer 1999). In Hemophilia B, the FIX gene is 33.5 kb long, has eight exons and codes for the FIX factor, a single chain glycoprotein (Yoshitake et al. 1985). As most of the exons in the Hemophilia B gene are relatively short, direct sequencing of a complete exon is possible once it has been amplified from genomic DNA with the PCR technique.
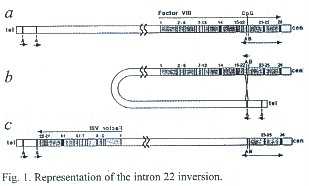
Mutations at both genes exhibit extreme diversity, in most cases they are point mutations. Sequence analysis of FVIII and IX-genes to detecte the precise nature of any mutation is expensive, time consuming and it is offered only by a few specialized centres. Therefore many laboratories indirectly assess the genetic status of potential carriers using genetic markers linked to the FVIII- or FIX-genes respectively (Giannelli et al. 1984, Herrmann et al. 1990, Harper et al. 1984, Peake 1992). Such markers are e.g. restriction fragment length polymorphism (RFLPs), short tandem repeats (STS) or variable number tandem repeats (VNTRs) which can be identified by Southern blotting or the polymerase chain reaction (PCR). In about 5% of the patients with severe Hemophilia deletions of the factor VIII/IX-genes can be detected by Southern blotting (Herrmann et al. 1990, Peake 1992).
The Costa Rican population has recently surpassed the 4.0 millions inhabitants (INEC 2002-CCP). At present there are 130 families with Hemophilia A and 30 families with Hemophilia B registered. Currently, the diagnosis is performed in the country only by standard laboratory tests and only one hospital offers specialized care and treatment to patients. In this article we present the molecular results of five families, three with Hemophilia A and two with Hemophilia B.
Material and methods
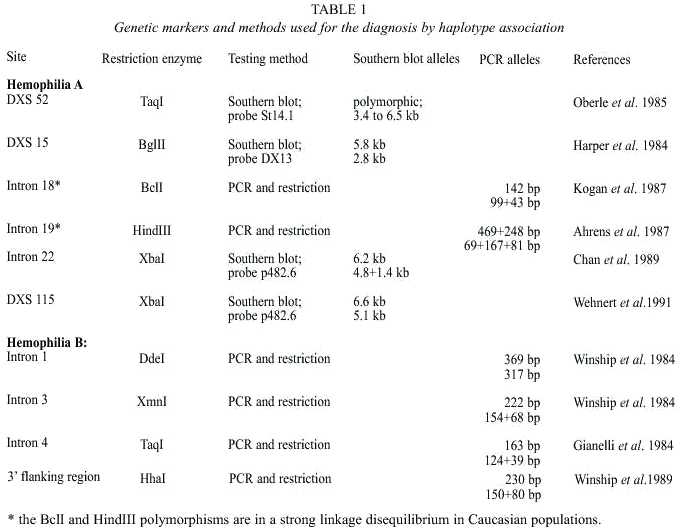
DNA Preparation and Genotyping: DNA was prepared by standard methods from whole blood (Miller et al. 1988). For the indirect genomic diagnosis of Hemophilia A and B several common RFLP have been characterized by Southern blotting and PCR (Table 1) (Giannelli et al. 1984, Peake 1992, Ahrens et al. 1987, Chan et al. 1989, Kogan et al. 1987, Oberle et al. 1985, Wion et al. 1986, Wehnert et al. 1990, Wehnert et al. 1991, Winship et al. 1984, Winship et al. 1989). The inversion in FVIII was detected by Southern blotting after restriction with BclI and hybridization with probe p482.6 (Harper et al. 1984). The deletion in FIX gene was detected by restriction of the total DNA with the enzymes TaqI and EcoRI respectively and hybridization with a cDNA probe of factor IX (Giannelli et al. 1984, Herrmann et al. 1990, Winship et al. 1989). For mutation analysis all exons of the FIX gene have been amplified by PCR (Wulff et al. 1995). The PCR products were screened by heteroduplex analysis. Small mutations (<30bp) in the FIX gene were made evident by shifted extra bands. Mutations in exons a and h do not lead in all cases to visible shift bands in the heteroduplex analysis or single-strand conformation polymorphisms (SSCPs) (Winship et al. 1984). Mutation analysis of the FIX gene was also performed by PCR and cycle sequencing procedure using Taq Dye Deoxy-terminator kit and the 373 ABI sequencer (Wulff et al. 1995, Herrmann et al. 1998, Wulff et al. 1999).
Subjects: From June 2000 to June 2002 data and blood samples of 5 Costa Rican families including patients and their relatives were collected. Females relatives of Hemophilia A patients showed an FVIII activity >75%. All participants gave informed consent according to a protocol approved by the University of Costa Rica.
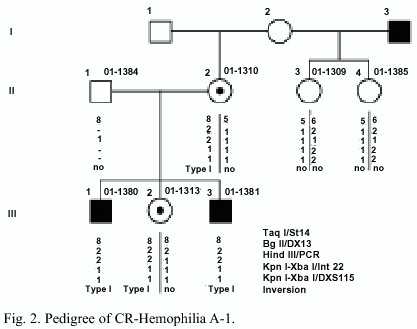
Family HA-1: Four female relatives of the patients with severe Hemophilia A asked for genetic counselling (Fig. 2). The patients (III-1; III-3) are treated prophylactically at home.
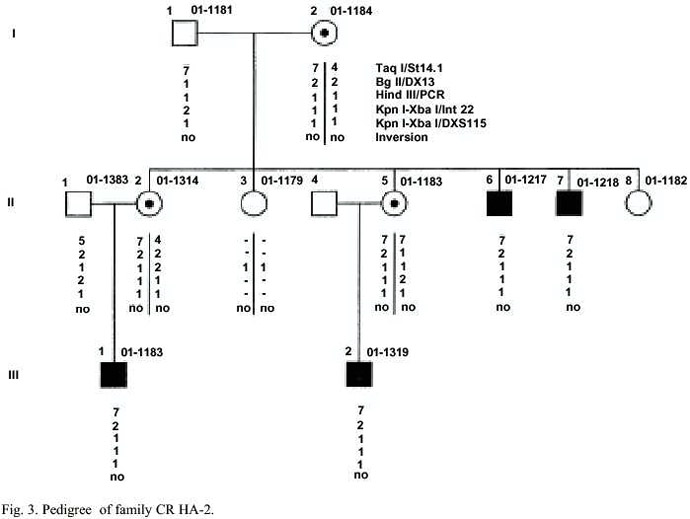
Family HA-2: In this family a severe Hemophilia Awith factor VIII activity of <1% is evident. Patients II-6; II-7; III-1 and III-2 (Fig. 3) suffered severe bleedings and they are treated prophylactically at home. Four females relatives of the patients asked for genetic counselling.
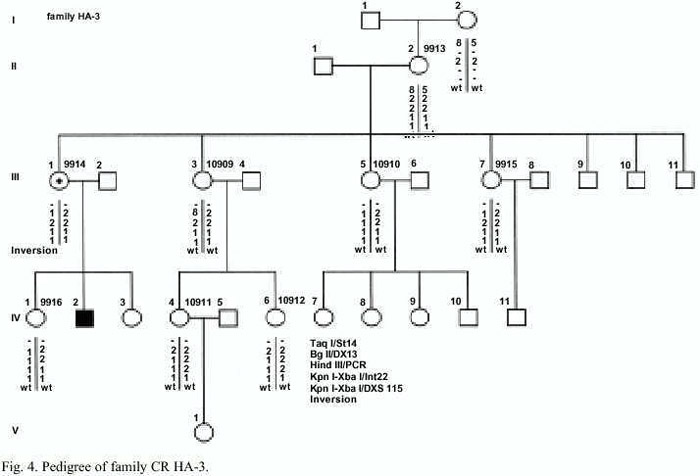
Family HA-3: Nine female relatives of an isolated patient (IV-2) with severe Hemophilia A asked for genetic counselling (Fig. 4). The patient is treated prophylactically at home. The patients DNA was not available for molecular analysis.
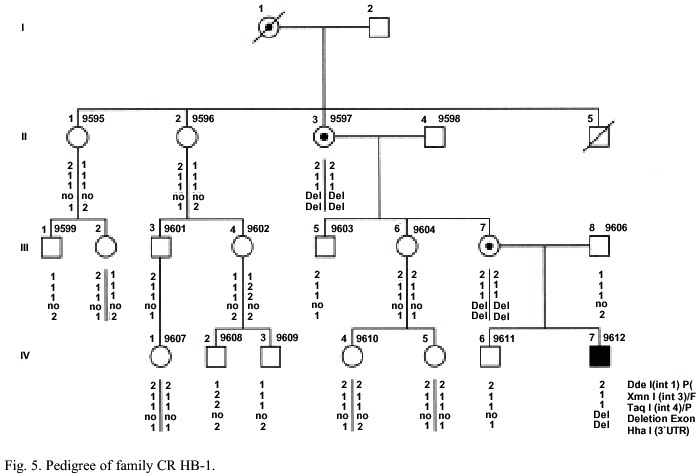
Family HB-1: The 7 year old patient IV-7 has a FIX activity <1%. Joint bleedings in knees, elbows and calves were evident. Recently, the patient is at prophylactic home treatment with prothrombinic complex. Ten females at risk to be a carrier asked for diagnosis (Fig. 5). The II-3 and III-7 females had a FIX activity of near 35% and presented moderate bleeding events after surgical treatment and tooth extraction.
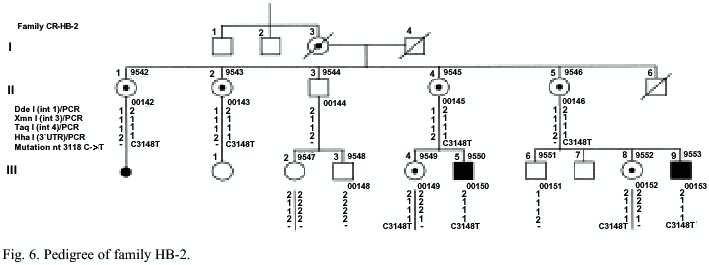
Family HB-2: In this family two Hemophilia B patients and seven females at risk to be a carrier asked for diagnosis . Both patients III-5 and III-9 (Fig. 6) have a severe phenotype with an FIX activity <1%. Patient III-5 suffered from severe bleedings in the subdural region with PCI and epilepsy. Recently, he got at prophylactic treatment at hospital twice a week.
Results
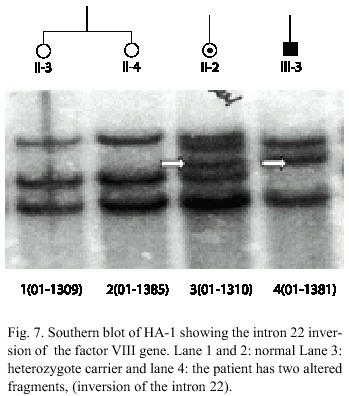
Family HA-1: The Hemophilia A in this family is caused by an inversion type I in the F VIII gene (Fig. 7). In this family the inversion of the intron 22 could be detected in the mother II-2, one daughter III-1 and in the patients (Fig. 2). The inversion enables a sure carrier detection and prenatal diagnosis in this family.
Family HA-2: The inversion is absent in patients of this family. Thus, three intergenic and two intragenic RFLPs have been characterized for indirect diagnosis. By pedigree analysis it is clear that the females I-2, II-2 and II-5 are obligate carries for Hemophilia A, (Fig. 3). I-2 is heterozygote only for the intergenic RFLP Taq I/St14.1. With this RFLP for the daughters II-2 and II-5 a carrier diagnosis can be offered with a diagnostic risk of about 2-5%. In case of pregnancy, II-2 a prenatal diagnosis can be offered base on the RFLP TaqI/St14.1 and the intragenic RFLP Hind III (PCR) and for II-5 on the intragenic RFLP KpnI-Xba I-int22. Both RFLPs enable an indirect diagnosis with a diagnostic risk of 0.1%.
Family HA-3: An inversion type I in the factor VIII gene causes the Hemophilia A. In this family the inversion of the intron 22 could be detected only in the mother III-1 (Fig. 4). Therefore she is a carrier of Hemophilia A. The inversion enables a sure carrier detection and prenatal diagnosis in this family. In the DNA of the females I-2, II-2, III-3, III-5, III-7, IV-1; IV-4 and IV-6 the inversion is not present, they can be excluded as a carriers.
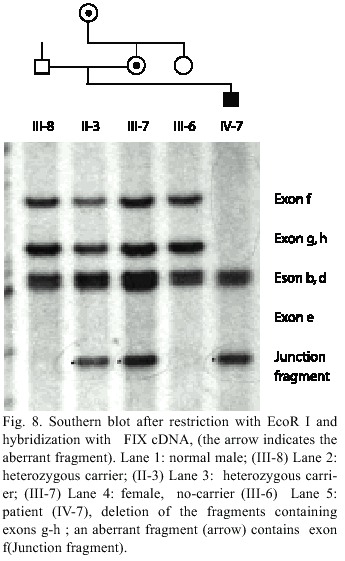
Family HB-1: Southern blot analysis after restriction with the enzymes EcoRI and TaqI respectively and hybridization with a factor IX cDNA showed that one fragment typical for the exons g and h is missing. The EcoRI – blot showed a smaller aberrant fragment (Fig. 8). The region downstream of the FIX gene with the HhaI polymorphism cannot be amplified. This result indicates a deletion of exons g and h, which extends more than 8 kb downstream of the FIX gene in the patients DNA. An aberrant restriction (junction) fragment include the exon f of the FIX gene can used for the direct diagnosis, for the detection of female carriers in this family (II-1 and III-7). A prenatal diagnosis can be offered to the carriers IV-7.
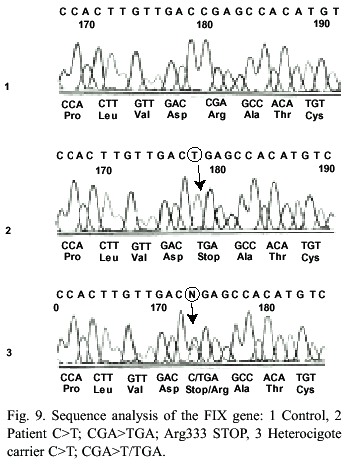
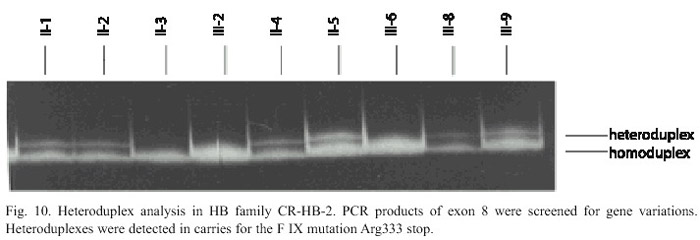
Family HB-2: In this family the causative mutation was analysed by amplification of all exons, the exon/intron regions and the promoter of the FIX gene. The PCR products of all exons were screened for variations by heteroduplex analysis. In PCR products of exon 8 of patients III-5 and III-9 and the females II-1, II-2, II-4, II-5, III-4 and III-8 were found shift bands using heteroduplex analysis. By sequencing of exon 8 of the patients DNA the nonsense mutation Arg333stop in nucleotide 31118C->T was found (Fig. 9). In the DNA of the females II-1, II-2, II-4, II-5, III-4 and III-8 the FIX mutation 31118C>T has been detected by heteroduplex analysis (Fig. 10) and sequencing (Fig. 9). They all are carriers for Hemophilia B. The mutation could not be detected in the DNA of proband III-2. She can be excluded as a carrier. In case of a pregnancy all carriers of this family a prenatal diagnosis could be offered by mutation analysis of fetal DNA. By indirect diagnosis II-4 and II-5 the possibility of a prenatal diagnosis also could be offered. The carrier status of II-1 and II-2 would not have been identified by indirect diagnosis. A prenatal diagnosis just for the detection of the linkage group not linked to the Hemophilia would be possible.
Discussion
The detection of carriership of Hemophilia A by the conventional methods has a probability between 5% and 95%, the main limitation of these methods is that they examine the phenotype (level of the factor) rather than the genotype (DNA) (Furier and Furier 1990, Herrmann et al. 1990). In fact, the results are probabilistic because the higher overlap existing between the activity levels according the lionization phenomenon. In Hemophilia B, carrier detection is also complicated for this reason and the molecular heterogeneity (Chistolini et al. 1990). The molecular technology has provided the possibility for exactly carrier analyses and prenatal diagnosis as precondition for genetic counselling (Herrmann and Scharrer 1999). In case that the mutation leading to the Hemophilia A or B is not known or the direct mutation detection is not possible, the indirect diagnosis or heteroduplex analysis is alternatives for the carrier detection in families at risk (Wulff et al. 1995, Wulff et al. 1997, Wulff et al. 1999).
In our study, we described the results of fifty-six subjects (9 patients) belong to five CR Hemophilia families and fourteen carriers were detected. Two Hemophilia A families, present the inversion in the intron 22 of the FVIII gene, which is a common mutation and it is described as the main cause (40-50%) for severe cases of Hemophilia A (Lakich et al. 1993). One family was not possible to detected this mutation in this gene, so in this case was useful the indirect genomic diagnosis, we used extragenic and intragenic probes. In order to track the individual copies of the FVIII gene in heterozygotes, it is essential to have available polymorphisms, which allow differentiating between the two copies present in female. The presence of heterozygosity or "informativeness" is essential for the indirect diagnosis. For the FVIII gene there are two types of polymorphisms, which segregate close with this gene (Chistonilli et al. 1990). Additionally to the most frequent biallelic polymorphisms resulting from single nucleotide substitutions, which create or abolish restriction sites (RFLPs), multiallelic CA repeat sequences are known in the FVIII-gene (Herrmann et al. 1990, Peake 1992). Using these genomic markers about 95% of females at risk a diagnosis can be offered, as in this Hemophilia A family.
We detected a deletion of exon g and h of the FIX gene and a nonsense mutation as a cause of the severe disease in the two Hemophilia B families. They are known mutations related with development of inhibitors, that is a complication in the hemophilic treatment (Wulff et al. 2003). The indirect diagnosis, the heteroduplex analysis and sequenciacion of the FIX gene were utilized for detecting these mutations. In the heteroduplex, PCR products of the different exon regions were analysed by gel electrophoresis. Mutation in the FIX gene becomes evident by heteroduplex bands. This method can be used for mutation screening, for carrier detection and prenatal diagnosis. A disadvantage of this method is that not all mutations cause shift bands, which are detectable under standard conditions (Wulff et al. 1997).
In conclusion, we showed in this study, the importance of the improvement of the molecular analysis for the Costa Rican Hemophilia, the utility of the direct and indirect diagnosis for the molecular characterization in the patients and the possibility of carrier detection and prenatal diagnosis for the females.
Acknowledgment
This work was made possible by the support from the Faculty of Medicine, Ernst-Moritz-Arndt- University, Greifswald Germany; Deutscher Akademischer Austauschdienst (DAAD) and by Vicerrectoría de Investigación, University of Costa Rica (project N. 807-A0-163).
Resumen
La hemofilia A y B es una enfermedad hemorrágica hereditaria ligada al cromosoma X, producida por la deficiencia del factor VIII o IX, respectivamente. Los individuso afectados presentan un fenotipo de hemorragia variable causada por el amplio espectro de mutacionesdentro del gen del factor VIII o IX. Se reportan los resultados preliminares del diagnóstico molecular de familias hemofilicas costarricenses. Se demuestran los hallazgos obtenidos por medio de diagnóstico molecular directo e indirecto en tres familias con hemofilia A y dos con hemofilia B; así como las precondiciones y facilidad de este diagnóstico. En dos familias con hemofilia A y dos con hemofilia B, la mutación responsable pudo ser detectada por medio de Southern Blot, por la reacción en cadena de la polimerasa o por secuenciación genética. Una familia con hemofilia A pudo ser analizada solamente por medio de análisis indirecto por medio de marcadores genéticos intragénicos y extragénicos.
References
Ahrens, P., T.A. Kruse, M. Schwarty, P.B. Rasmussen & N. Din. 1987. A new HindIII restriction fragment length polymorphism in the Hemophilia A locus. Hum. Genet. 76: 127-128. [ Links ]
Chan, V., T.N. Tong & T.P. Chan. 1989. Multiple XbaI polymorphisms for carrier detection and prenatal diagnosis of haemophilia A. Br. J. Haematol. 73: 497-500. [ Links ]
Chistolini, A., M. Papacchini, M.G. Mazzucconi, G. La Verde, R. Arcieri, A. Ferrari, R. Paesano, A. Pachi & G. Mariani. 1990. Carrier detection and prenatal diagnosis in Haemophilia A and B. Hematology 75: 424-428. [ Links ]
Forbes, C.D., L.M. Aledort & R. Madhok. 1997. Hemophilia. Chapman Hall Medical. London. pp. 3-143. [ Links ]
Furie, B. & B. Furie. 1990. Hemophilia basis of Hemophilia. Sem. Hematol. 27: 270-285. [ Links ]
Giannelli, F., D.S. Anson, K.H. Choo, D.J. Ress, P.R. Einship, N. Ferrari, C.R. Rizza & G.G. Brownlee. 1984. Characterization and use of an intragenic polymorphic marker for detection of carriers of haemophilia B (factor IX deficiency). Lancet 1 (8371): 239-241. [ Links ]
Gitschier, J., W.I. Wood, T.M. Goralka, K.L. Wion, E.Y. Chen, D.H. Eaton, G.A.Vehar, D.J. Capon & R.M. Lawn. 1984. Characterization of the human factor VIII gene. Nature 312: 326-330. [ Links ]
Harper, K., M.E. Pembrey, R.M. Winter, D. Hartley & E.G.D. Tuddenham.1984. A clinically useful DNA probe closely linked to haemophilia A. Lancet 2 (8393): 6-8. [ Links ]
Herrmann, F.H. & I. Scharrer.1999. Humangenetische Beratung bei Hämophilie A und B. pp. 3-33. In Deutsche Hämophiliegesellschaft (eds.). Mitteilungen der Deutschen Hämophiliegesellschaft zur Bekämpfung von Blutungskrankheiten, Hamburg. [ Links ]
Herrmann, F.H., M. Wehnert, W. Schröder & K. Wulff. 1990. Genomic diagnosis of Hemophilia A and B. Thromb. Haemorrh. Disorders 2(1): 11-15. [ Links ]
Herrmann, F.H. & K. Wulff. 1998. Molekulare Genanalyse und Geniagnostik bei Hämophilie B und FVII Mangel. Hämostaseologie 3: 129-139. [ Links ]
INEC-CCP. Costa Rica: estimaciones y proyecciones de población actualizadas al año 2000. Período 1970- 2050. San José, Instituto Nacional de Estadística y Censos y Centro Centroamericano de Población, Universidad de Costa Rica. (En prensa). [ Links ]
Kogan, S.C., M. Doherty & J. Gitschier. 1987. An improved method for prenatal diagnosis of genetic diseases by analysis of amplified DNA sequences. Application to Hemophilia A. N. Engl. J. Med. 317: 985-990. [ Links ]
Lakich, D., H. Kazazian, S.E. Antonarakis & J. Gitschier. 1993 Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nature Genet. 5: 236-241. [ Links ]
Miller, S.A., D.D. Dykes & H.F. Polesky. 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res. 16: 1215. [ Links ]
Oberle I., G. Camerino, R. Heilig, L. Grunebaum, J.P. Cazenave, C. Crapanzano, P.M. Mannucci & J.L. Mandel. 1985. Genetic screening for haemohilia A with a polymorphic DNA probe. N. Engl. J. Med. 312: 682-686. [ Links ]
Pattinson, J.K., D.S. Millar, J.H. McVery, C.B.Grundy, K. Wieland & R.S. Misbashan. 1990. The molecular genetic analysis of Hemophilia A: A directed search strategy for the detection of point mutation in the human factor VIII gene. Blood 76: 2242-2248. [ Links ]
Peake, I. 1992. Registry of DNA Polymorphisms within or close to the Human Factor VIII and IX Genes. Thromb. Haemost. 67: 277-280. [ Links ]
Wehnert, M., W. Schröder & F.H. Herrmann. 1991. Two new markers at DXS 115 useful for genomic diagnosis in haemophilia A. Thromb. Haemorrh. Disorders 3(1): 21-24. [ Links ]
Wehnert, M., E.L. Shukova, V.I. Surin, W. Schroder, Solovjev Gya & F.H. Herrmann. 1990. Prenatal diagnosis of haemophilia by the polymerase chain reaction using the intragenic Hind III polymorphism. Prenatal Diagnosis 10: 529-532. [ Links ]
Winship, P.R., D.S. Anson, C.R. Rizza & G.G. Brownlee. 1984. Carrier detection in haemophilia B using two futher intragenic restriction fragment length polymorphisms. Nucleic Acid Res. 12: 8861-8872. [ Links ]
Winship, P.R., D.J. Rees & M. Alkan. 1989. Detection of polymorphisms at cytosine phosphoguanadine dinucleotides and diagnosis of haemophilia B carriers. Lancet 1(8639): 631-634. [ Links ]
Wion, K.L., E.G.D. Tuddenham & R.M. Lawn. 1986. A new polymorphism in the factor VIII gene for prenatal diagnosis of haemophilia A. Nucleic Acid Res. 14: 4535-4542. [ Links ]
Wulff, K., K. Bykowska, S. Lopaciuk & F.H. Herrmann. 1999. Molecular analysis of Hemophilia B in Poland: 12 novel mutations of the factor IX gene. Acta Biochim. Polonica. 721-726. [ Links ]
Wulff, K., U. Ebener, Ch-S. Wehnert, P.A. Ward, U. Reuner, W. Hiebsch, F.H. Herrmann & M. Wehnert. 1997. Direct molecular genetic diagnosis and heterozygote identification in X-linked Emery-Dreifuss muscular dystrophy by heterodeplex analysis. Dis. Markers 13: 77-86. [ Links ]
Wulff, K., W. Schröder, M. Wehnert & F.H. Herrmann. 1995. Twenty-five novel mutations of the factor IX gene in haemophilia B. Human Mutat. 6: 346-348. [ Links ]
Wulff, K., W. Schröder, M. Wehnert & F.H. Herrmann. 2003. Molecular Analysis of Hemophilia B: Greifswald Registry FIX deficiency (HemophiliaB). pp.35-48 In I.III. Scharrer, W. Schramm (eds.). 32nd Hemophilia Symposium Hamburg 2001. Springer, Berlin. [ Links ]
Yoshitake, S., B.G. Schach, D.C. Foster, E.W. Davie & K. Kurachi. 1985. Nucelotide sequence of the gene of human factor IX ( haemophilic factor B). Biochem. 24: 3736-3750. [ Links ]

