










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.52 n.3 San José Sep. 2004
Disabilities caused by unstable mutations in Costa Rica
Patricia Cuenca1,2,3 *, Fernando Morales1,3,4 & Isabel Castro1,4
1 Sección de Genética Humana, Instituto de Investigaciones en Salud (INISA). Universidad de Costa Rica, 2060, San José, Costa Rica.
2 Escuela de Biología. Universidad de Costa Rica, 2060, San José, Costa Rica.
3 Programa Institucional de Neurociencias. Universidad de Costa Rica, 2060, San José, Costa Rica.
4 Escuela de Medicina. Universidad de Costa Rica, 2060, San José, Costa Rica.
* Phone: 506-224 3668; Fax: 506-207 5130; pcuenca@cariari.ucr.ac.cr
Received 24-V-2004. Corrected 23-VII-2004. Accepted 24-VIII-2004.
Abstract
Myotonic dystrophy and fragile X syndrome are two genetically determined relatively common disabilities. Both are examples of a new type of mutation mechanism called unstable or dynamic mutations, triple repeats expansions or DNA amplification. Fragile X syndrome is recognized as the main cause of hereditary mental retardation and myotonic dystrophy is considered the most common muscular dystrophy of adults. This is a prospective non randomized study of clinically affected people, in order to confirm the diagnosis with molecular techniques (Southern blot and PCR) and to perform cascade screening of the rest of the family to offer them adequate genetic counseling. We were able to corroborate the initial diagnosis in most clinical cases of myotonic dystrophy, but in the cases of mental retardation more than half studies were negative for fragile X syndrome, stressing the difficulties encountered by medical practitioners to diagnose this syndrome. The reasons for this are several; probable the main culprit is the subtle and unspecific clinical picture affected individuals exhibit, particularly children before puberty. Cascade screening, genetic counseling and selective abortion are the only tools available to prevent these disabling diseases for the moment. Rev. Biol. Trop. 52(3): 501-505. Epub 2004 Dic 15.
Key words: unstable mutations, fragile X syndrome, myotonic dystrophy, molecular diagnsosis, Costa Rica.
Palabras clave: mutaciones inestables, síndrome del cromosoma X frágil, distrofia miotónica, diagnóstico molecular, Costa Rica.
Unstable mutations are responsible for an increasing number of genetic neurological diseases since 1991 (Cuenca and Morales 1999). This recently discovered kind of mutation arises when three adjacent nucleotides are repeated in tandem in numbers greater than normal and thus alter the actual gene function, their RNAs or proteic products, and consequently cause neurological or systemic disorders. Diseases of this origin and of higher prevalence are fragile X syndrome (FRAXA) – the main cause of hereditary mental retardation, affecting approximately 6% of severely mentally retarded persons, predominantly males (Castro 1998) - and myotonic dystrophy type 1 (DM1) - the most common muscular dystrophy of the adult (Harper 1998). The molecular defect underlying DM is the expansion of an unstable (CTG)n trinucleotide repeat in the 3 untranslated region of a protein kinase gene, DMPK, on chromosome 19q13.3 (Buxton et al. 1992). In DM 1, unaffected people have from 5 to 37 repeats, but affected people have from 50 to several thousands repeats. The genetic basis for FRAXA was revealed by positional cloning in 1991 and resulted in an expansion of the triplet CGG in the 5 untranslated region in the FMR1 gene (fragile X mental retardation-1) localized near the distal boundary of Xq27.3 (Kremer et al. 1991). In FRAXA, unaffected people have from 7 to 59 repeats, patients with premutation have from 60 to 230 repeats and affected patients (with the full mutation) have more that 230 repeats. Both diseases are disabling and have anticipation in common, meaning increasing severity of disabilities in subsequent generations in FRAXA, and both increasing severity and earlier age of onset in the case of muscular dystrophy type 1 (Morales et al. 2001, Cuenca et al. 2002).
Both diseases segregate as dominants and thus many affected persons are found in these families. Treatment for DNA amplification diseases is far from reach, and the only way to avoid occurrence or recurrence in the families is through contraception or prenatal diagnosis and selective abortion. In either case, prevention of disease can be achieved if adequate genetic counseling is given at the right time to at risk members. A prerequisite for genetic counseling is the precise diagnosis of the molecular status of unaffected but carrier family members. It is for this reason we have the objective to perform molecular diagnosis of these unstable mutations, not only to confirm the clinical impression, but to investigate as many family members as possible to offer them the adequate genetic counseling that will enable them to choose the best reproductive option they can have.
Materials and methods
This is a prospective non randomized study of clinically affected people, in order to confirm the diagnosis with molecular techniques and to perform cascade screening of the rest of the family to offer them adequate genetic counseling. In the mental retardation cases the category of the FMR1 alleles (normal, premutation or full mutation) is assessed as well as the number of CGG repeats in premutation carriers and the methylation status of the 5´ UTR of the gene. Southern blot was performed using probe StB12.3, restriction enzymes Hind III or EcoRI and EagI to determine methylation status, according to Rousseau et al. (1991). Normal bands are of 5.2 or 2.8Kb and 5.2Kb respectively (Fig. 1). PCR was used to refine the diagnosis and performed according to Chong et al. (1994). In cases of clinical suspicion of myotonic dystrophy type 1, the protocol implies quantification of the number of CTG repeats at the 3 non coding end of the DMPK gene. This was performed following procedures of Shelbourne et al. (1992) (Fig. 2). Genetic counseling was performed according to international standards, with plenty of educational materials and taking into account the socioeconomic and cultural status of the patients.


Results
For fragile X syndrome 93 index cases of mental retardation and relatives were studied, distributed as follows: (1) From 11 families (more than two related persons), 71 individuals were studied. In five families the unstable mutation was confirmed, with 15 individuals with mental retardation and full mutation, another 15 individuals had the premutation and so were normal carriers, and 16 had normal amount of repeats. In six families 25 individuals were studied and proved to have normal alleles. One of these families came to us with cytogenetic diagnosis of fragile X syndrome in the four mentally retarded offspring. (2) In two brother-brother pairs, one pair of first cousins and another mother-son duo, all eight individuals resulted with normal alleles. (3) Finally, in 14 mentally retarded children referred for molecular study, only one girl resulted with a full mutation.
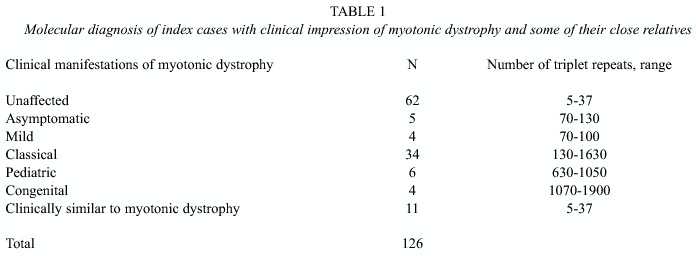
For muscular dystrophy cases, 126 molecular diagnoses were performed as follows: (1) We studied 104 individuals from 18 families. In four families with 27 index cases and relatives studied, myotonic dystrophy diagnosis was discarded since all alleles were normal. In 14 families 77 individuals were studied and the results are shown in table 1. (2) In six pairs of individuals, one sister-sister, two mother-daughter and three mother-son, we found the mutation in three pairs. In this group there are two cases of pediatric onset, one case of classical presentation, another case of Steinerts disease (congenital onset) and two individuals free of mutation. (3) Ten clinical cases of myotonic dystrophy were studied and eight proved positive with the classical manifestation of the disease and two resulted with normal alleles.
Discussion
These results highlight the different predictive value of clinical diagnosis in these two diseases in the three subgroups studied, much higher for myotonic dystrophy and not so good for fragile X syndrome. The former disease is often referred to the neurologist, specially the most affected cases, and the signs and symptoms are highly suggestive of the correct diagnosis, when a good quality electromyography and physical test is performed. However, the DM phenotype is often misleading, and differential diagnosis is sometimes hard work for the physicians, due to the similarity with other myotonic conditions, and sometimes some patients are misdiagnosed as DM1. The accurate diagnosis however, can be achieved only at the molecular level. A clinical diagnosis of DM1 but negative at the molecular level could indicate the presence of other myotonic conditions, such as: DM2 or Cl- or Na+ channelopathies. For fragile X syndrome, on the contrary, the clinical picture is highly unspecific, particularly in prepuberal children and most of them are classified as cases of mental retardation of unknown origin. This is true not only in Costa Rica, but in more developed countries as well. Moreover, of mental retardation cases showing X linkage, fragile X syndrome accounts for no more than half of them. This is a relatively new disease for most medical practitioners, the clinical picture besides from being subtle is blurred for most of them and so they either ignore the disease or refer all cases of mental retardation for molecular study. Even cytogenetic diagnosis of index cases can be misleading and false negative or false positive results are not rare, on the contrary, false negatives arise from faulty lymphocyte culture methods and false positives from common fragile sites near the rare one associated with fragile X syndrome at Xq27.3. This last situation might explain the family with four mentally retarded children which where positive on cytogenetic grounds but we found negative at the molecular, more accurate level. Furthermore, we studied these children for FRAXE mental retardation and were negative as well. Cytogenetic diagnosis is less accurate and time consuming, and so we have abandoned this method in favor of molecular studies in index cases and relatives, but fifteen years ago it was the only test we had and allowed us to report a 6% prevalence of fragile X syndrome in a population of mentally retarded children attending a special education school (Castro and Cuenca 1987, 1996).
According to the practical experience of at least four studies that have attempted cascade screening for fragile X syndrome, it has proved to be feasible and useful. Follow up of the screened positive families has evidenced a marked reduction in the birth of affected children through contraception and prenatal diagnosis (Murray et al. 1997).
In both genetic diseases studied, the accurate diagnosis of the unstable mutation associated to each of them allowed unambiguous detection of clinically affected individuals and of healthy carriers. In this way, we were able to demonstrate an erroneous clinical or cytogenetic diagnosis in several disabled individuals, who consequently had to be studied for other etiology for their diseases. Even more important was the detection of healthy carrier individuals at high risk for disabled offspring. These people received genetic counseling in order to help them take the best reproductive options according to their own risk and harm perceptions, moral and religious beliefs, economic capability to afford the expense of rehabilitation of affected family members and other issues.
We consider of utmost importance to perform molecular diagnosis in cases with clinical impression of disability associated to unstable mutations in genes FMR1 and DMPK in Costa Rica. To increase the level of suspicion in medical practitioners it is necessary for them to acknowledge the constant need to keep in pace with genetic advances, particularly of these two relatively common diseases.
The correct identification of index cases allows cascade screening which in turn enables adequate genetic counseling to the families at risk and the possibility of prevention of these diseases in Costa Rica. Since these disabilities pose a considerable burden to the caretakers in particular and to society in general, our health authorities must support and finance our molecular genetic projects on behalf of our population.
Acknowledgments
To Federico Hernández and Fernando Ortiz from INISA; to the neurologists Roberto Brian, Mauricio Sittenfeld and Gerardo del Valle; to Regina Neumann, Harmut Peters and Peter Nürnberg from Charité, Berlin; to Peter Steinbach from Ulm University; to Keith Johnson and Darren Monckton from Glasgow University; to Tetsuo Ashizawa and Alberto Rosa from Baylor College of Medicine; to the International Atomic Energy Agency; the British Embassy in Costa Rica; the DAAD and to the Vicerrectoría de Investigación of the University of Costa Rica.
Resumen
La distrofia miotónica tipo1 (DM1) y el síndrome del cromosoma X frágil (FRAXA) son dos enfermedades hereditarias relativamente comunes. Ambas constituyen ejemplos de un nuevo tipo de mecanismo mutacional, llamado mutaciones inestables o dinámicas, expansión de tripletas, o amplificación del ADN. La DM1 se considera como la distrofia muscular más frecuente en los adultos y FRAXA es la principal causa de retardo mental hereditario. Este trabajo presenta resultados actualizados de un estudio prospectivo no aleatorio en pacientes clínicamente afectados, que se realiza con el objetivo de confirmar el diagnóstico con técnicas moleculares (Hibridación de Southern y reacción en cadena de la polimerasa, PCR), y llevar a cabo el tamizaje en cascada del resto de la familia para ofrecerles consejo genético adecuado. Se confirmó el diagnóstico clínico inicial en la mayoría de los casos de distrofia miotónica, pero en los casos con retardo mental, más de la mitad de los análisis resultaron negativos para la amplificación en el gen FMR1, específica de FRAXA. La razón principal para esto podría ser el cuadro clínico muy sutil que muestran los niños afectados antes de la pubertad. Los únicos métodos disponibles para prevenir estas discapacidades por el momento son, el tamizaje en cascada, el consejo genético y el aborto selectivo. De los cuales, el último no se puede llevar a cabo según las leyes vigentes en Costa Rica.
References
Buxton, J., P. Shelbourne, J. Davies, C. Jones, T. Van Tongeren, C. Aslanidis , P. De Jong, G. Jansen, M. Anvret, B. Riley, R. Williamsom & K. Johnson. 1992. Detection of an unstable fragment of DNA specific to individuals with DM. Nature 355: 547-548. [ Links ]
Castro, I. 1998. Definición, clasificación, etiología, diagnóstico y prevención del retardo mental. Acta Méd. Cost. 40: 7-14. [ Links ]
Castro, I. & P. Cuenca. 1987. Tamizaje de sitio frágil en el cromosoma X en una población de retardados mentales. Hallazgos preliminares. Rev. Méd. Hosp. Nal.. Niños Costa Rica 22: 11-14. [ Links ]
Castro, I & P Cuenca. 1996. Frecuencia del síndrome del cromosoma X frágil en la Escuela de Enseñanza Especial "Fernando Centeno Güell". Acta Ped. Cost. 10: 99-106. [ Links ]
Chong, S., E. Eicher, M. Hughes & D. Nelson. 1994. Robust amplification of the fragile X syndrome CGG repeat using Pfu polymerase: ethidium bromide detection of normal and premutation alleles. Am. J. Med. Genet. 51: 522-526. [ Links ]
Cuenca, P. & F. Morales. 1999. Mutaciones inestables, causa de algunas enfermedades neurológicas hereditarias. Acta Méd. Cost. 41: 7-15. [ Links ]
Cuenca, P., F. Morales & I. Castro. 2002. Diagnóstico directo de la mutación que causa el síndrome del cromosoma X frágil. Experiencia en Costa Rica. Acta Méd. Cost. 44: 27-33. [ Links ]
Harper, P. 1998. Myotonic dystrophy as a trinucleotide repeat disorder - a clinical perspective. pp. 115-130. In R.D. Wells & S.T. Warren (eds.). Genetics Instabilities and hereditary neurological diseases. Academic, San Diego, California. [ Links ]
Kremer, E., M. Pritchard, M. Lynch, S. Yu, K. Holman, E. Baker, S.T. Warren, D. Schlessinger, G.R. Sutherland & R.I. Richard. 1991. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252: 1711- 1714. [ Links ]
Morales, F., P. Cuenca, R. Brian-Gago, M. Sittenfeld-Appel & G. del Valle-Carazo. 2001. Diagnóstico molecular de la distrofia miotónica (DM) en Costa Rica. Acta Méd. Cost. 43: 159-167. [ Links ]
Murray, J., H. Cuckle, G. Taylor & J. Hewison. 1997. Screening for fragile X syndrome: information needs for health planners. J. Med. Screen. 4: 60-94. [ Links ]
Rousseau, F., D. Heitz , V. Biancalana, S. Blumendfeld, C. Kretz , J. Boué, N. Tommerup, C. Van der Hagen, C. De Lozier-Blanchet, M.F. Croquette, S. Gilgenkrantz, P. Jalbert, M.A. Voelckel, I. Oberlé & J.L. Mandel. 1991. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 325: 1673-1681. [ Links ]
Shelbourne, P. , R. Wingvist, E. Kunert, J. Davies, J. Leisti, H. Thiele, H. Bachmann, J. Buxton, B. Williamson & K. Johnson. 1992. Unstable DNA may be responsible for the incomplete penetrance of the myotonic dystrophy phenotype. Hum. Mol. Genet. 1: 467-473. [ Links ]

