











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkRevista de Biología Tropical
versión On-line ISSN 0034-7744versión impresa ISSN 0034-7744
Rev. biol. trop vol.48 no.2-3 San José jun. 2000
María Virginia Solís 1
Recibido 8-VII-1999. Corregido 25-XI-1999. Aceptado 1-XII-1999.
Abstract
The Lafora disease is an uncommon genetic condition. Four cases (two families) were detected in Zarcero, a small town in Costa Rica (population under 2000). They belonged to two separate consanguineous marriages but both families had common ancestors. The diagnosis of Lafora disease was confirmed by liver biopsy in one of the patients. The ages of onset were 13, 14, 16 and 17 years. Patients died after four, nine, six and five years of severe progressive physical and mental deterioration, respectively. The gene for Lafora disease arrive to Zarcero from one of its founders. There are no other cases reported from Costa Rica: this is an example of genetic drift, or more specifically, founder effect.
Key words
Lafora disease, myoclonic epilepsy, founder effect.
La enfermedad de Lafora fue descrita primero por Lafora (1911b) y Lafora y Glueck (1911) y consiste en un trastorno progresivo degenerativo del sistema nervioso central, de herencia autosómica recesiva. La mayor parte de los casos descritos han sido producto de matrimonios consanguíneos y la incidencia sexual es igual (Footitt et al. 1997). Es una enfermedad fatal rara caracterizada por convulsiones, mioclonías, deterioro mental progresivo, signos piramidales, cerebelares y extrapiramidales, ataxia, demencia y cuerpos de inclusión esféricos intracelulares positivos a la reacción periódica de ácido-Schiff (PAS) en varios tejidos (Blandfort et al. 1987, Vital et al. 1995). Estos depósitos de inclusión fueron descubiertos por el neuropatólogo Gonzalo Rodríguez Lafora en la enfermedad que hoy día lleva su nombre (Lafora 1911a, b, Fernández et al. 1997), consisten en polisacáridos ramificados (poliglucosanos) y la glucosa es el único azúcar componente (Yokota et al. 1988).
El método preferido para confirmar el diagnóstico de la enfermedad es la biopsia de piel, por medio de la cual se demuestra la presencia de los cuerpos de Lafora (Carpenter y Karpati, 1981, Thom y Revesz, 1996). No se ha informado acerca de anormalidades en la piel de los padres de los pacientes (Busard et al. 1987).
La enfermedad comienza al final de la niñez o durante la adolescencia (6-20 años) y conduce a un desenlace fatal dentro de los siguientes diez años de aparición de los primeros síntomas (Blandfort et al.1987, Vital et al. 1995). En la literatura se han descrito algunos pocos casos que han presentado la enfermedad a una mayor edad de la usual (Kaufman et al. 1993, Footitt et al. 1997).
Por medio de estudios de ligamiento genético y de mapeo por homocigosis se logró mapear el gen responsable de esta enfermedad, en el brazo largo del cromosoma 6: 6q23-q25(Serratosa et al. 1995). Un informe posterior lo restringió a 6q24 (Maddox et al. 1997, Sainz et al. 1997). Luego se identificó en 6q24 al gen EPM2A, el cual codifica una proteína con actividad de tirosina fosfatasa. Análisis genéticos realizados revelaron que los pacientes eran homocigotas para mutaciones en EPM2, las cuales provocaban la pérdida de su función. Por lo tanto, la enfermedad de Lafora resulta por la inactivación a través de mutaciones de la actividad de tirosina fosfatasa, que puede ser importante en el control del metabolismo del glicógeno (Serratosa et al. 1999). Por otra parte se ha encontrado que los pacientes con enfermedad de Lafora muestran heterogeneidad genética (Minassian et al. 1999).
El objetivo de la presente investigación fue tratar de describir el cuadro clínico de los pacientes afectados, establecer la procedencia del gen mutante que condujo a esta epilepsia y determinar para cada uno de los miembros de la genealogía, la probabilidad de heredar el gen en forma homocigota o heterocigota. Esta última parte será objeto de otra publicación.
Materiales y Métodos
Para recopilar la información necesaria se recurrió a las siguientes fuentes: entrevista personal de individuos pertenecientes a la genealogía, entrevista telefónica, envío de cartas, consulta de archivos parroquiales (Alfaro Ruiz, Naranjo, Grecia, Moravia, Santa Ana, Guadalupe, San Isidro de Coronado, Alajuela y de la Curia Metropolitana de San José), búsqueda de algunos datos (Registro Civil, Archivo Nacional y en el Museo Histórico de Juan Santamaría), entrevistas a parte de los médicos que atendieron a los pacientes, revisión de los expedientes hospitalarios y de la autopsia practicada a JEVS, búsqueda de los Cuerpos de Lafora en tejidos preservados de JEVS.
Se elaboró una genealogía parcial para tratar de establecer el origen ancestral del gen que produjo la enfermedad, así como genealogías para cada una de las familias relacionadas. Una vez que se logró ese objetivo, se hizo una gran genealogía en la cual se registraron únicamente los matrimonios consanguíneos ocurridos a través de varias generaciones. Lo anterior con la finalidad de detectar a otras posibles familias con riesgo de tener hijos afectados.
Resultados
La enfermedad apareció en Zarcero (10°11N; 84°23W), Alajuela (9°49–11°05N; 84°10-85°27W), Costa Rica, el cual según el censo de 1964, contaba con aproximadamente 1863 habitantes (Anónimo 1965). Se presentó un caso dentro de una familia de cuatro hijos (JEVS) y tres en otra de diez hermanos (AVSV, JASV, FASV) (Fig.1 y Cuadro 1).
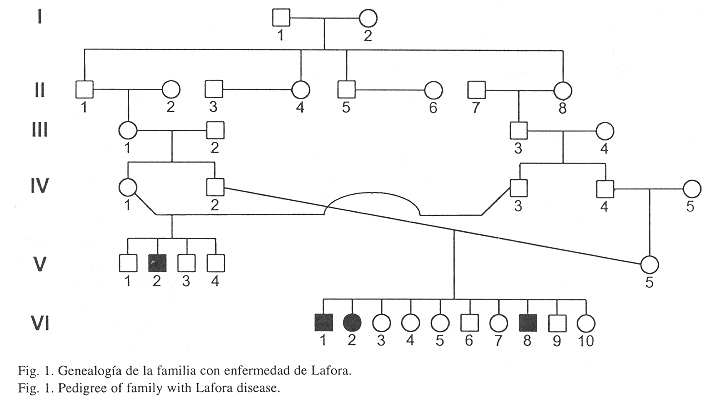
Hubo coincidencia entre las características de la enfermedad presentadas por los pacientes y las descritas en la literatura (Blandfort et al. 1987).
Hay un tronco familiar común para ambos matrimonios (Fig.1). Con los datos disponibles no fue posible determinar cuál de los dos individuos I-1 o I-2 era portador del gen mutante, pero sí que la probabilidad de sus hijos de heredar el gen respectivo era de 0.5.
Unicamente a uno de los afectados (JEVS) se le realizó autopsia, ya que murió estando hospitalizado. Se identificó los cuerpos de Lafora en una biopsia de hígado, conservada desde hacía veinticinco años.
CUADRO 1.
Algunas características de la enfermedad de Lafora en los individuos estudiados.
TABLE 1.
Some characteristics of Lafora disease in the studied patients.
| | | | |
| | |||
| | | | |
| | | | |
| | | | |
| | | | |
Discusión
Zarcero, es un pueblo autosuficiente. Eso ha favorecido el matrimonio entre familiares ya que la mayor parte de sus habitantes descienden de ocho familias fundadoras, que llegaron alrededor de 1854 (Anónimo 1965).
I-1 y I-2 contrajeron matrimonio en el centro de la capital de San José en 1833 y I-2 murió en Zarcero en 1912. II-1 y II-2 se casaron en Moravia, San José en 1866 y II-1 falleció en Zarcero en 1923. II-7 y II-8 se desposaron en Moravia, San José en 1862 y II-8 murió en Zarcero en 1942. Esos datos demuestran que ellos formaron parte de las primeras familias que habitaron Zarcero.
Según la investigación genealógica, II-1 y II-8 heredaron el gen de la Enfermedad de Lafora, de alguno de sus padres. Varias generaciones después, cuando ocurrieron esos dos matrimonios consanguíneos, el gen alcanzó su estado homocigota y apareció la enfermedad. Se detectaron más matrimonios entre miembros de los descendientes de I-1 y I-2, pero no se han presentado más casos.
La Enfermedad de Lafora es una dolencia rara a nivel mundial y en Costa Rica no hay datos de otros casos. Por lo tanto, el presente informe constituye un ejemplo del efecto fundador (Griffiths et al. 1996), por medio del cual el gen mutante llegó a alcanzar una frecuencia alta en esta pequeña población y permitió su manifestación.
Los casos costarricenses tuvieron los síntomas de la enfermedad de Lafora clásica (Vital et al. 1995). Se documentó la presencia de los cuerpos de Lafora sólo en uno de los pacientes, pero los cuadros clínicos y su origen ancestral común indican que se trata de la misma dolencia.
Al igual que en esta investigación, en muchas familias con más de un hijo afectado hubo uniones consanguíneas (Footitt et al. 1997, Sainz et al. 1997).
El estudio genealógico detectó muchos individuos con riesgo de poseer el gen. Dado que el gen responsable de la enfermedad ya fue mapeado, es probable que en un futuro cercano, dispongamos de la tecnología molecular necesaria para discriminar a los heterocigotas y de esta manera poder brindarles consejo genético.
Agradecimientos
Agradecemos a la Vicerrectoría de Investigación, Universidad de Costa Rica, por el apoyo económico y logístico, así como a Thais Fonseca, por su ayuda, al patólogo Minor Vargas por confirmar la enfermedad y a la neuróloga Genoveva Molina por su asistencia en la interpretación de la información clínica.
Resumen
La enfermedad de Lafora es una condición genética infrecuente. Cuatro casos (dos familias) fueron detectados en Zarcero, una pequeña localidad de Costa Rica (población bajo 2000). Ellos pertenecieron a dos matrimonios consanguíneos y ambas familias tenían ancestros comunes. El diagnóstico de Enfermedad de Lafora fue confirmado por medio de una biopsia de hígado en uno de los pacientes. Las edades de aparición fueron 13, 14, 16 y 17 años. Los pacientes murieron después de cuatro, nueve, seis y cinco años de deterioro físico y mental severo, respectivamente. El gen de la enfermedad de Lafora llegó a Zarcero a partir de uno de sus fundadores. No han habido informes de otros casos en Costa Rica: este es un ejemplo de deriva genética, específicamente el efecto fundador.
Referencias
Anónimo. 1965. Monografía del Cantón de Alfaro Ruiz. Imprenta Nacional, San José, 64p. [ Links ]
Blandfort, M, T. Tsuboi & F. Vogel. 1987. Genetic counseling in the epilepsies. Hum. Genet. 76: 303-331. [ Links ]
Busard, H.L., A.A. Gabreëls-Festen, W.O. Renier & A.M. Stadhouders. 1987. Axilla skin biopsy: a reliable test for the diagnosis of Lafora's disease. Ann. Neurol. 21: 599-601. [ Links ]
Carpenter, S. & G. Karpati. 1981. Sweat gland duct cells in Lafora disease: Diagnosis by skin biopsy. Neurology 31: 1564-1568. [ Links ]
Fernández, V., J.M. Moreno, A. Martín, M.L. García, B. Revilla &J.L. Moreno. 1997. Lafora and neuropathology. Rev. Neurol. 25: 2036- 2039. [ Links ]
Footitt, D.R., N. Quinn, R.S. Kocen, B. Oz & F. Scaravilli. 1997. Familial Lafora disease of late onset: report of four cases in one family and a review of the literature. J. Neurol. 244: 40- 44.
Griffiths, A.J., J.H. Miller, D.T. Suzuki, R.C. Lewontin & W.M. Gelbart. 1996. An introduction to genetic analysis. W. H. Freeman and Company, New York, 916p. [ Links ]
Kaufman, M.A., A.J. Dwork, N.J. Willson, S. John & J.D. Liu. 1993. Late-onset Lafora's disease with typical intraneuronal inclusions. Neurology 43: 1246-1248. [ Links ]
Lafora, G.R. 1911a. The presence of amyloid bodies in the protoplasm of the ganglion cells: a contribution to the study of the amyloid substance in the nervous system. Bull. Gov. Hosp. Insane 3: 83- 92. [ Links ]
Lafora, G.R. 1911b. Über das Vorkommen amyloider Körperchen im Innern der Ganglienzellen: zugleich ein Bietrag zum Studium der amyloiden Substanz im Nervensystem. Virchows Arch. 205: 295-303. [ Links ]
Lafora G.R. & B. Glueck. 1911. Beitrag zur Histopathologie der myoklonischen Epilepsie. Z.Gesamte Neurol.Psych. 6: 1-14. [ Links ]
Maddox, L.O., M. Descartes, J. Collins, J. Keating, S. Rosenfeld, C. Palmer, A.J. Carroll & R. Kuzniecky. 1997. Identification of a recombination event narrowing the Lafora disease gene region. J. Med. Genet. 34: 590-591. [ Links ]
Minassian, B.A., J. Sainz, J.M. Serratosa, M. Gee, L.M. Sakamoto, S. Bohlega, G. Geoffroy, C. Barr, S.W. Scherer, U. Tomiyasu, n S.Carpenter, K. Wigg, A.V. Sanghvi & A.V. Delgado-Escueta. 1999. Genetic locus heterogeneity in Laforas progressive myoclonus epilepsy. Ann. Neurol. 45: 262-265. [ Links ]
Sainz, J., B.A. Minassian, J.M. Serratosa, M.N. Gee, L.M. Sakamoto, R. Iranmanesh, S. Bohlega, R.J. Baumann, S. Ryan, R.S. Sparkes & A.V. Delgado-Escueta. 1997. Lafora progressive myoclonus epilepsy: narrowing the chromosome 6q24 locus by recombinations and homozigosities. A. J. Hum. Genet. 61: 1205-1209. [ Links ]
Serratosa, J.M., A.V. Delgadoescueta, I. Posada, S. Shih, I. Drury, J., Berciano, J.A. Zabala, M.C. Antunez & R.S. Sparkes. 1995. The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum. Molec. Genet. 4: 1657- 1663. [ Links ]
Serratosa, J.M., P. Gomez-Garre, M.E. Gallardo, B. Anta, D.B. de Bernabe, D. Lindhout, P.B. Augustijn, C.A. Tassinari, R.M. Malafosse, M. Topcu, D. Grid, C. Dravet, S.F. Berkovic, S.R. de Cordoba. 1999. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum. Mol. Genet. 8: 345-352. [ Links ]
Thom, M. & T. Revesz. 1996. Typical polyglucosan bodies are present in the sweat gland lumina in Laforas disease. Acta Neuropathol. 92: 102-103. [ Links ]
Vital, A., J. Lamarche, J.P. Bernier, P. Boulan-Predseil, H. Loiseau, A. Rougier, P. Henry & C. Vital. 1995. Signification des inclusions de polymères de glucose en neuropathologie. Ann. Pathol. 15: 269-275. [ Links ]
Yokota, T., T. Ishihara, H. Yoshida, M. Takahashi, F. Uchino & S. Hamanaka. 1988. Monoclonal antibody against polyglucosan isolated from myocardium of a patient with Lafora disease. J. Neuropathol. Exp. Neurol. 47: 572-577. [ Links ]
1 Escuela de Biología, Universidad de Costa Rica, San José, Costa Rica. Fax (506)207-4216. vsolisa@cariari.ucr.ac.cr

