










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Costarricense
versión On-line ISSN 0001-6002versión impresa ISSN 0001-6012
Acta méd. costarric vol.63 no.2 San José abr./jun. 2021
Clinical case
Double heterozygous patient for New York Hemoglobin (HB Kaohsiung) and Alpha Thalassemia (-α3.7/ααα)
Double heterozygous patient for New York Hemoglobin (HB Kaohsiung) and Alpha Thalassemia (-α3.7/ααα)
1Caja Costarricense de Seguro Social, CCSS, Laboratorio Clínico Área de Salud de Aserrí.Costa Rica
2 Caja Costarricense del Seguro Social, CCSS, Hospital Nacional de Niños Dr. Carlos Sáenz Herrera, Laboratorio de Tamizaje Neonatal y Alto Riesgo. Costa Rica
3 Caja Costarricense del Seguro Social, CCSS, Hospital Nacional de Niños Dr. Carlos Sáenz Herrera, Laboratorio de Estudios Especializados e Investigación. Costa Rica
Hemoglobinopathies are common in the tropics, being particularly frequent in heterozygous carriers of hemoglobin S (HbS) and alpha thalassemias. Alpha thalassemias are diseases with autosomal recessive inheritance patterns, characterized by microcytic hypochromic anemias of variable intensity. 1,2
Hemoglobin New York (HbNY) or Kaohsiung hemoglobin is a variant of hemoglobin that occurs by mutation at amino acid 114 in the beta-globin chain, modifying a valine by glutamic acid, and was first identified in 1967 in a family of Chinese descent.3
Individuals heterozygous for hemoglobin New York are clinically normal without hematologic alteration.4,5 However , severe complications have been described in double heterozygotes for HbNY and Hemoglobin S (HbS), which has been described as a severe form of sickle cell disease and is associated with complications such as retinopathy, and spleen infarction. 6
If hemoglobin New York is inherited together with a deletion of 3 alpha genes, it could lead to severe Hb H disease. For its correct diagnosis, it is important to have at least two different methodologies for the detection of hemoglobin variants, if only one method is used such as high-pressure liquid chromatography (HPLC) the Hb NY variant could go undetected,7 since it runs at the same position as normal hemoglobin A. In this particular case, as the sample was processed by the capillary electrophoresis method, an electrophoretic peak could be detected in the position where Hb NY normally runs.
In Costa Rica, even though patients with Hb NY have been previously described;8,9 the high prevalence of carriers for HbS and Thalassemia makes the finding of a double heterozygote of hemoglobin NY/ -3.7 Alpha Thalassemia in the Aserrí Health Area relevant and merits follow-up, to provide the patient with a correct interpretation of their hematometric indices and timely genetic counseling.
Presentation of the case
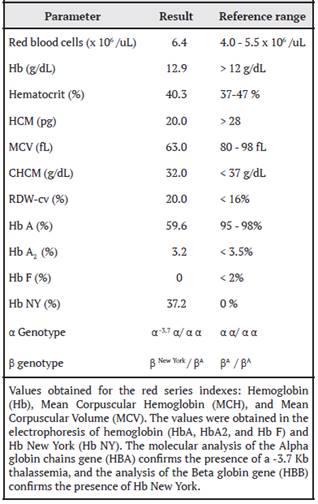
A 40-year-old white woman of Guanacaste ancestry, asymptomatic, with no family history of anemia or chronic diseases, presents to the Clinical Laboratory of Aserrí for routine follow-up of anemia. The red bloodcount indices such as hemoglobin (Hb), Mean Corpuscular Volume (MCV) and Mean Corpuscular Hemoglobin (MCH) are not as expected for the Red Blood Cell count obtained, which is compatible with thalassemia (Table 1). The leukocyte and platelet counts are within the reference range.
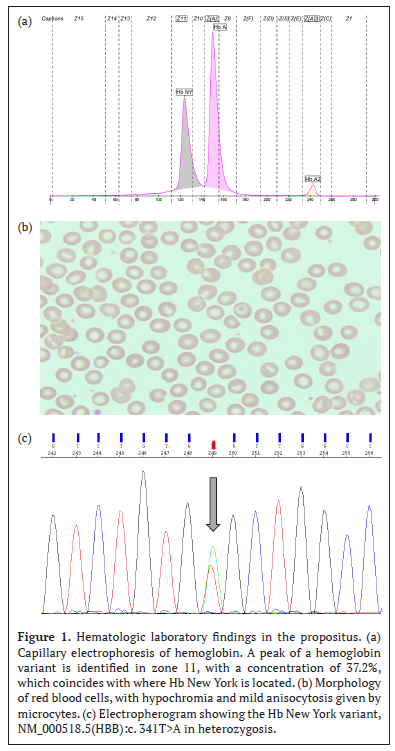
When reviewing the patient’s medical record, it is evident that the hemacytometric indices lead the treating physician to follow up for iron deficiency anemia. However, when using the diagnostic algorithm of the Clinical Laboratory, these values together with the morphology of red blood cells (Figure 1b), lead to suspect a thalassemia syndrome, so a peripheral blood sample is referred to the Laboratory of Specialized Studies and Research (LEEI) of the National Children’s Hospital (HNN) Dr. Carlos Sáenz Herrera, for study by hemoglobin electrophoresis.
Hemoglobin electrophoresis is performed by capillary electrophoresis (Capillarys 2; Serbia, Paris, France; software version 6.2). The normal value obtained for HbA2 rules out the presence of Beta-thalassemia, and the value obtained for fetal hemoglobin (HbF) rules out the presence of Delta Beta-thalassemia. But the presence of an abnormal peak hemoglobin concentration of 37.2% in zone 11 suggests the presence of a hemoglobin variant known as Hemoglobin New York (Figure 1a). It is noteworthy that the concentration of HbNY is lower than what is observed in heterozygous patients but concentrations similar to those reported in cases of double heterozygotes HbNY and Alpha Tal -3.7.7
For the molecular analysis for Alpha Thalassemia, whose methodology consists of the identification of 21 of the most frequent mutations/deletions of the α-globin gene based on PCR amplification and reverse hybridization (ViennaLab labor-diagnostikaGmbH, Vienna, Austria); genomic DNA was extracted from the peripheral blood leukocytes of the propositus and its progenitors. By PCR and reverse hybridization, the heterozygous -3.7 mutation was identified.
The National Neonatal and High-Risk Screening Laboratory performed amplification of the coding regions with specific primers for the HBB gene and subsequently sequenced by capillary electrophoresis (Sanger Sequencing, AB3500 from Thermo Fisher Scientific) using BigDye® Terminator v3.1 reagents (Thermo Fisher Scientific). Sequencing analysis detected the variant NM_000518.5(HBB):c. 341T>A, which changes the amino acid sense, where the valine at position 14 is replaced by glutamic acid (GTG>GAG, p.(Val114Glu)); and is considered pathogenic causing the New York or Kaohsiung hemoglobin phenotype (Figure 1c).
Discussion
Hb New York is very common in southern China,8 and is the most frequent hemoglobin variant after Hb E, followed by Hb Q Thailand in third place, so its detection in Asia is common, but not in our country.9 Heterozygous genotypes for hemoglobin New York are asymptomatic without hematological alteration,9 but in association with other hemoglobinopathies such as alpha or beta thalassemias or hemoglobin S, the individual manifests more severe pictures.6 The identification of these variants represents an important finding that allows the clinician to give a correct interpretation of the hemacytometric indices, to offer the patient an adequate clinical follow-up, and genetic counseling, and to know the possible implications for their offspring.
In the case of our patient, there is no anemia or chronic disease in her family history. The patient was being followed up for iron deficiency anemia which did not respond to treatment.
Hence the importance of applying a diagnostic algorithm that allows the correct interpretation of all hemacytometric indices and red blood cell morphology, which allowed the identification of double heterozygosity for alpha thalassemia and Hb NY as the cause of microcytic anemia that did not respond to treatment.
In our country, there are very few reported cases of heterozygotes for Hb NY, and in the analysis of national publications, we found no records of any previous cases of double heterozygotes for Hb NY and Alpha Thalassemia -3.7 kd, this being the first case described in the Costa Rican scientific literature. Because of the clinical implications described above, genetic counseling is a priority, and the patient was made aware of this. Acknowledgments: To the graduate assistant Keren Vanegas García, of the Clinical Laboratory of the Aserrí Health Area for her contributions to the technical processing of the sample.
Referencias
1. Flint J, Harding R, Boyce A, JB Clegg. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1998; 11: 1-51 [ Links ]
2. Piel F, Weatherall D. The alfa-thalasemias. NEJM. 2014; 371: 1908-1916. [ Links ]
3. Ranny H, Jacobs A, Nagel R. Haemoglobin New York. Nature. 1967; 213: 876-879 [ Links ]
4. Panyasai S, Pornprasert S. Detection of Hemoglobin New York (b113 (G15) Val-Glu,GTG>GAG) in a Thai Woman by Capillary Electrophoresis. Indian J Hematol Blood Transfus. 2016; 32:514-516 [ Links ]
5. Chaibunruang A, Singha K, Srivorakun H, Fucharoen G, Fucharoen S. Molecular Characteristics of Hb New York (b113 (G15) Val-Glu,GTG>GAG) in Thailand. Hemoglobin. 2018; 42:1-5 [ Links ]
6. McFarlane A, Warkentin TE, Cartin W. A novel sickling hemoglobinopathy. NEJM. 2011; 365:1548-1549 [ Links ]
7. Dong-Zhi L. Hemoglobin New York is not a matter in prenatal screening and diagnosis for β-thalassemia. Ped Hematol Oncol. 2008; 25:769-771 [ Links ]
8. Saenz GF, Elizondo J, Arroyo G. Two cases of hemoglobin New York in Costa Rica. Hemoglobin. 1980; 4: 101-105 [ Links ]
9. Sáenz-Renauld GF. Hemoglobinas anormales. Acta méd. Costarric. 2005; 47:173-179 [ Links ]
Received: October 15, 2020; Accepted: March 24, 2021
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
