









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkLas alfa talasemias son las enfermedades monogénicas más comunes en el ser humano, poseen un patrón hereditario autosómico recesivo, cursan con anemias microcíticas hipocrómicas de intensidad variable, cuya clínica va desde casi asintomáticos (en el caso de portadores) hasta anemias hemolíticas que podrían poner en riesgo la vida del paciente.1 En las alfa talasemias se afecta cuantitativamente la producción de las cadenas de globina alfa(α), y ocurren a una frecuencia mayor que las betatalasemias, estimando que el 5% de la población mundial es portadora de la variante -4.2 de alfatalasemia.1 En el adulto sano, la Hb está conformada por un 95 % a un 98% de hemoglobina A (HbA, α2β2); menos del 1% de hemoglobina fetal (HbF, α2ϒ2), y menos del 3,5% de hemoglobina A2 (HbA2, α2δ2). Los genes que codifican para las cadenas alfa y gamma están duplicados (αα/αα; ϒϒ/ϒϒ), mientras que los genes de las cadenas beta están codificados por un locus simple (β/β). Se han identificado más de 100 genotipos de alfa talasemia, con fenotipos que varían desde los asintomáticos (genotipos silentes) hasta las formas letales (como el síndrome de hydrops fetalis ), correlacionándose la severidad con el número de copias no funcionales del gen de la α-globina. Según el número de genes afectados, las alfa talasemias se clasifican en dos subgrupos: las α+-talasemias (antes llamadas α-talasemia2), en las cuales uno de los alelos de uno o ambos genes está delecionado o inactivado por mutaciones concretas (-α/αα; -α/-α; ααND/αα) y las α0-talasemias (antes llamadas α-talasemia1), en las cuales ambos alelos del mismo gen de α-globina son delecionados en un mismo cromosoma (--/αα).
En el adulto, el exceso de las cadenas β forma un tetrámero (β4), conocido como hemoglobina H (HbH). La enfermedad por HbH se caracteriza por presentar cuadros de anemia hemolítica de intensidad variable. Los casos clínicamente relevantes de enfermedad por HbH, usualmente involucran una alfa+-talasemia y una alfa0-talasemia coheredada (-α/-- o ααND/--), es decir, un doble heterocigota. Los casos que resultan de deleciones, suelen tener una presentación clínica leve, con moderada anemia y leve esplenomegalia, pocos periodos de infecciones y por lo general no requieren transfusiones sanguíneas. Un ejemplo de este tipo es la variante sudeste asiático (--SEA/αα), común en países asiáticos. Los cuadros clínicos más severos se producen con las variantes no delecionales, como con la variante de hemoglobina Constant Spring (HbCS), la cual también es muy común en países asiáticos,2 en cuyo caso se presenta un cuadro clínico con crisis hemolíticas asociadas a estrés oxidativo desde las primeras etapas de la vida, esplenomegalia, sobrecarga de hierro, mayor frecuencia de infecciones, úlceras en miembros inferiores, litiasis y deficiencia de ácido fólico.3,4
En el feto, la producción defectuosa de las cadenas alfa (α) se refleja por la presencia de un exceso de cadenas gamma (ϒ), las cuales forman un tetrámero (ϒ4), conocido como hemoglobina Bart (HbBart). La presencia de HbBart en recién nacidos indica afectación de uno o más de los cuatro genes de alfa globina, lo que causa alfa talasemia. Dado que los patrones electroforéticos de las talasemias usualmente cursan normales para los portadores, el diagnóstico de las alfa talasemias solo puede ser confirmado por análisis molecular del ADN. Los estudios moleculares han permitido detectar temprano gran cantidad de heterocigotos por alfa talasemia, los cuales no se hubiesen podido detectar basando el diagnóstico solo en presencia de HbBart en el recién nacido.2 En Costa Rica, el programa de tamizaje neonatal incluyó la detección de variantes de Hb y estudios por hemoglobinopatías desde 2005,5 lo que permite que el niño sea valorado de forma oportuna por el Servicio de Hematología del Hospital Nacional de Niños (HNN) “Dr. Carlos Sáenz Herrera”, cuando es detectada alguna variante de hemoglobina en la prueba de tamizaje. La siguiente descripción de caso pretende dar a conocer algunos de los fenotipos y genotipos de alfa talasemia presentes en la población de Costa Rica, cuya importancia radica en ser el primer caso descrito en el país que evidencia genotipo compuesto sudeste asiático y hemoglobina Constant Spring.
Presentación del caso
Se describe aquí el primer caso en Costa Rica de dos hermanos con enfermedad por hemoglobina H, dobles heterocigotos para (- -SEA/ααCS). El propositus 1 corresponde a una paciente femenina de 4 años y 10 meses de edad, nacida pretérmino a las 35 semanas de edad gestacional. Fue valorada inicialmente al mes de vida en un hospital regional, por antecedente de ictericia prolongada, Hb en 6,5 g/dL, Coombs directo negativo, requirió transfusión de glóbulos rojos empacados, que no corrigió la anemia, por lo que fue referida al Servicio de Hematología del HNN a los dos meses de vida. Al momento de ingreso al HNN la madre indica el antecedente heredo-familiar de hermano con talasemia de quien, al no tener control en el Servicio de Hematología, se desconocía su condición de portador o talasémico. Durante el tamizaje neonatal de esta paciente se tomaron dos muestras, las cuales fueron analizadas con cromatografía de alta resolución en el programa de Tamizaje Neonatal; ambas sugirieron la presencia de alfa talasemia. El hemograma inicial de ingreso del propositus 1 en el HNN mostró Hb 6,7g/dL, GR 3,27 x 106/uL, VCM 71,3 fL, HCM 20,9 pg y RDW 22 %. La morfología de glóbulos rojos mostró moderada hipocromía, moderada anisopoiquilocitosis con microcitosis, basofilia difusa y reticulocitos en 6,46 %. Los demás parámetros del hemograma eran normales. Se transfunde una segunda vez a los dos meses de vida en el HNN y no requirió más transfusiones. Se realiza electroforesis de Hb por electroforesis capilar (Capillarys2; Sebia, París, Francia; software versión 6.2) y se observa presencia de HbH, HbBart y de un pico en la zona C donde se ubica la variante de HbCS, a una concentración del 2,3 %, por lo que se realiza estudio molecular por alfa talasemia. En la Figura 1 se muestran las morfologías de glóbulos rojos y electroforesis de hemoglobina de las muestras control de ambos propositus.
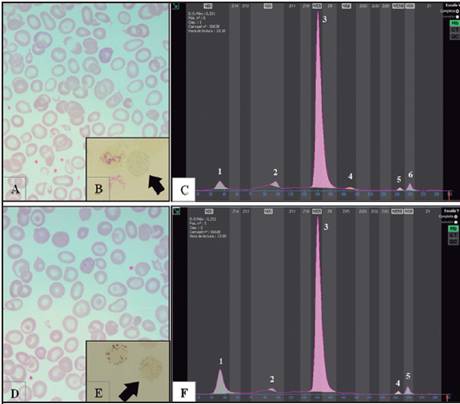
Figura 1 Hallazgos hematológicos de laboratorio de los propositus. (A) Morfología de glóbulos rojos, propositus 1. (B) Cuerpos de inclusión, propositus 1. (C) Electroforesis capilar de Hb, propositus 1, (1: HbH, 2: HbBart, 3: HbA, 4: HbF, 5: HbA2, 6: HbCS). (D) Morfología de glóbulos rojos, propositus 2. (E) Cuerpos de inclusión, propositus 2. (F) Electroforesis capilar de Hb, propositus 2, (1: HbH, 2: HbBart, 3: HbA, 4: HbA2, 5: HbCS)
El propositus 2 corresponde a un masculino de 8 años, quien inició control en el HNN a la edad de 6 años, debido a que su hermana fue referida al Servicio de Hematología del HNN, con enfermedad por hemoglobina H. La morfología de glóbulos rojos permite evidenciar marcada anisopoiquilocitosis, con clara basofilia difusa. Al nacimiento, el tamizaje neonatal de este paciente se había reportado con patrón electroforético FA + Bart (mediante cromatografía por isoelectroenfoque), lo que sugiere enfermedad por hemoglobina H, sin embargo, en ese momento, se le realizó una sola muestra, pues no fue posible localizarlo para tomar la segunda. Al ingreso en el HNN, en la electroforesis de hemoglobina se obtiene un patrón A/H/ Bart (cuadro 1), y también se observa un pico ubicado en la zona C, con la variante HbCS a una concentración del 2,4%. No necesitó transfusiones previas ni posteriores al ingreso al HNN. Se realiza electroforesis de hemoglobina de ambos progenitores (cuadro 1).
Cuadro 1 Hallazgos del hemograma, electroforéticos y moleculares del propositus y sus progenitores
| Parámetro | Propositus 1 | Propositus 2 | Madre | Padre |
| Sexo | F | M | F | M |
| GR (x106/μL) | 5,06 | 5,12 | 5,74 | 5,88 |
| Hb (g/dL) | 9,2 | 9,0 | 12,3 | 15,3 |
| VCM (fL) | 59,3 | 57,4 | 63,6 | 76,5 |
| HCM (pg) | 18,2 | 17,6 | 21,4 | 26,0 |
| CHCM (g/dL) | 30,7 | 30,6 | 33,7 | 34,0 |
| HbA (%) | 88,5 | 82,4 | 97,6 | 97,0 |
| HbA2 (%) | 0,7 | 0,7 | 2,4 | 2,6 |
| HbF (%) | 1,2 | ND | ND | ND |
| HbH (%) | 5,0 | 13,5 | ND | ND |
| HbBart (%) | 2,3 | 1,0 | ND | ND |
| HbCS (%) | 2,3 | 2,4 | ND | 0,4 |
| Genotipo | --SEA/ααCS | --SEA/ααCS | --SEA/αα | αα/ααCS |
Hb: hemoglobina; GR: recuento glóbulos rojos; VCM: volumen corpuscular medio; HCM: hemoglobina corpuscular media; CHCM: concentración de hemoglobina corpuscular media; ND: no detectado; SEA: variante sudeste asiático; HbCS: variante de hemoglobina Constant Spring.
La madre presentó una morfología de glóbulos rojos con leve hipocromía, moderada anisopoiquilocitosis con microcitos, esferocitos, eliptocitos, esquizocitos, dacriocitos y codocitos. El padre tiene una morfología de glóbulos rojos normocítica normocrómica. Se descartó otras posibles causas del cuadro clínico de los propositus, tales como incompatibilidad de grupo madre-feto, las infecciones congénitas por TORCH (toxoplasma, rubeola, citomegalovirus y herpes virus), enfermedad cardiaca congénita, deficiencia de glucosa 6 fosfato deshidrogenasa, entre otras, con el fin de hacer diagnóstico diferencial.
Luego se realizan ensayos moleculares, cuya metodología consiste en la identificación de 21 de las mutaciones / deleciones más frecuentes del gen α-globina, a partir de amplificación por PCR e hibridación inversa (ViennaLab). El ADN genómico fue extraído de los leucocitos de sangre periférica de los pacientes. En el Cuadro 1 se describen los hallazgos moleculares y de las electroforesis, tanto de los progenitores como de los propositus. En ambos niños se identifica la mutación del gen HBA2:c.427T>C (tipo HbCS) y deleción doble del gen tipo (-- SEA), cuyos resultados son compatibles con alfa talasemia doble heterocigota para (--SEA/ααCS). El estudio molecular de la madre resultó heterocigoto para deleción SEA (--SEA/αα). El padre es heterocigoto para la mutación (αα/ααCS). Cabe destacar que tanto la madre como el padre de los menores son de origen chino, y que la madre refiere que el padre era talasémico, desconociendo su condición genotípica y sus antecedentes clínicos antes de iniciar el estudio de los niños en el HNN.
Discusión
Los casos en estudio corresponden a dos hermanos doble heterocigotos para alfa talasemia, quienes presentan, concomitantemente, una mutación en uno de los genes de alfa globina que da lugar a la variante HbCS, y la deleción en el otro gen tipo sudeste asiático. Sus progenitores son de origen chino y dicho genotipo fue heredado de ellos, quienes poseen de igual manera el rasgo alfa talasémico, pero con carácter heterocigota.
La descripción de este caso toma relevancia dentro de la población de Costa Rica debido a que, a pesar de que nuestro país no posee una alta frecuencia de alfa talasemias, existe mucha migración (turistas y residentes), lo cual explica el hecho de que aparezcan genotipos talasémicos poco frecuentes en la población.
La mayor frecuencia a nivel mundial en la expresión del rasgo alfa talasémico se produce en zonas tropicales y subtropicales, incluso en algunas áreas como Sudeste de Asia, Oriente Medio y Mediterráneo, la frecuencia de portadores podría llegar a ser de hasta un 80 %.6,7Además, específicamente en las zonas de Tailandia, China y el Sudeste de Asia, la enfermedad por hemoglobina H posee una alta prevalencia.8 Con respecto a las variantes en estudio, la deleción SEA es la más común en asiáticos alrededor del mundo, y la talasemia asociada a la HbCS es la variante de alfa talasemia no delecional más común en China.9,7
Debido a la migración de las poblaciones, la situación endémica de las talasemias ha cambiado de manera dramática en los últimos años, y con esto algunos rasgos han sido encontrados globalmente en zonas con baja incidencia, pero con alta migración,6,7como es el caso de Costa Rica. Dado que la población migrante en Costa Rica es del 9,0 %, y que cerca del 0,2 % corresponde a población de origen chino (INEC, X Censo Nacional de Población y Vivienda, 2011) es factible que, en familias con ambos progenitores orientales residentes en nuestro país, se puedan presentar casos homocigotos o dobles heterocigotos para distintas variantes de alfa talasemia, lo que implicaría mayor probabilidad de detectar HbH en la población.
La herencia concomitante de ambos rasgos alfa talasémicos en los niños en estudio, hace que expresen un genotipo --SEA/ ααCS, derivado de la herencia del genotipo de la madre (--SEA/ αα) y del padre (αα/ααCS). Desde el punto de vista molecular, los genes que codifican para las cadenas alfa están duplicados (αα/αα), heredando un par de genes en cada cromosoma proveniente de cada progenitor. La deleción (--SEA) corresponde a una α0- talasemia y, por ende, de su herencia se obtienen dos genes afectados en el mismo cromosoma. Estudios de mapeo genético de la enfermedad por hemoglobina H han revelado la deleción de tres genes de α-globina en los individuos afectados (--/-α), y una proporción significativa de pacientes poseen concomitancia con la mutación HbCS. Estos pacientes con HbH y HbCS (--/ααCS) usualmente evidencian un grado más severo de anemia que pacientes con la presentación ordinaria de enfermedad por HbH (--/-α).8 La mutación que produce HbCS corresponde a una α+-talasemia, en cuyo caso se hereda un gen mutado y otro normal en el mismo cromosoma (ααCS).6 En los propositus del caso en estudio, al heredar un par de genes afectados con la deleción (--SEA) y otro con la mutación (ααCS), se posee un único gen funcional, y esto correlaciona con la presentación de cuadros de anemia hemolítica, pues la expresión de la enfermedad por HbH dependerá del número de copias no funcionales del gen de α-globina (--SEA/ααCS). Esto a su vez explica la aparición de variantes de hemoglobina como lo son la HbH y la HbBart, frecuentes en estos casos.6 Los genes α2 contribuyen de 1,5 a 3 veces más en la producción de cadenas de globina que los genes α1.6 La mutación que origina la HbCS se produce en el gen α2, lo que de nuevo correlaciona con la severidad de la presentación clínica de los niños, en los cuales al inicio se presenta como una anemia hemolítica que después logra cronicidad al disminuiir la velocidad de producción de cadenas α normales.6
La enfermedad por HbH y los casos de HbCS poseen características fenotípicamente opuestas, siendo estos últimos los que poseen un curso de enfermedad más severo cuando se presentan como genotipo homocigoto o en concomitancia con otras deleciones,7 condición que se manifiesta en ambos hermanos. A pesar de ello, la aparición de hemoglobinas con características electroforéticas distintas en las pruebas de tamizaje y en la electroforesis de hemoglobina (como la HbH o la HbBart) dirigen el diagnóstico, mas no es útil como diagnóstico temprano ni permite identificar portadores heterocigotos, razón por la cual el diagnóstico de las alfa talasemias solo puede ser confirmado por análisis molecular del ADN.1
Importancia del tamizaje por HbH
El cribado o tamizaje neonatal por alfa talasemia es particularmente útil en prevenir severas complicaciones maternas, como el síndrome hydrops fetalis por HbBart, y en establecer diagnósticos precisos en los casos en los que la alfa talasemia sea coheredada con hemoglobina S o beta talasemia, o incluso en los casos en los que se determine anemia por deficiencia de hierro.
En la bibliografía10 se han reportado dos casos de síndrome hydrops fetalis en pacientes dobles heterocigotos (--SEA/ααCS), con fenotipo similar al reportado en este caso clínico, de ahí la importancia de un diagnóstico temprano que permita incluso el control durante el embarazo y la consejería genética.
Como la mayoría de los programas de tamizaje neonatal poseen un enfoque preventivo, detectando principalmente beta talasemias y variantes de hemoglobina como la HbS en la drepanocitosis, no son adecuados para determinar la frecuencia de las alfa talasemias. En el caso de Costa Rica esta situación no es distinta de otros países, si bien es cierto nuestro programa de tamizaje neonatal es capaz de detectar la presencia de hemoglobina H y Bart, el diagnóstico definitivo de alfa talasemia requiere análisis molecular del gen alfa globina, que actualmente no es parte del programa de tamizaje neonatal. Por lo tanto, los programas de tamizaje neonatal deben considerar cualquier población en la que las variantes de alfa talasemia estén presentes, o en casos en donde no se pueda explicar la anemia microcítica hipocrómica en ausencia de deficiencia de hierro, refiriéndolos para su estudio por el Servicio de Hematología, tal y como se realiza en el Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”.
Con la globalización, el aumento de las poblaciones que se movilizan es ahora más frecuente, y la diversidad de las combinaciones de las variantes de alfa talasemias entre sí, o incluso con otras hemoglobinopatías, seguirá en aumento. Aun cuando el fenotipo exacto de tales combinaciones sea difícil de prever, conocer la distribución de las variantes genéticas al menos ayudará a definir los procedimientos diagnósticos para detectarlas. Si bien es cierto, las alfa talasemias no representan un problema clínico tan grande como las beta talasemias, las alfa talasemias son probablemente el desorden genético más común a nivel mundial y representan un modificador genético para otras condiciones, tales como la malaria, la drepanocitosis, las beta talasemias e incluso la anemia por deficiencia de hierro. Las alfa talasemias representan un problema de salud global, y por lo tanto, desde el punto de vista de salud pública, es importante conocer las variantes presentes en cada país o, en su defecto, estar atentos a la presencia de estas, provenientes de otras zonas geográficas de donde son comunes. Esto asegurará su correcto diagnóstico, ofrecer al paciente el tratamiento adecuado, y brindar el consejo genético correspondiente.