











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkI La activación inmune crónica constituye el desencadenante primario en la patogenia de la enfermedad por VIH.
Cuando se determinó que la infección por VIH estaba confinada a células CD4+ y tenía efectos citopáticos, se estableció la hipótesis de que el virus causaba inmunodeficiencia por eliminación directa de los linfocitos T CD4+ e impedimento en su recambio.1 Los mecanismos moleculares involucrados en la eliminación de las células T por parte del VIH han sido exhaustivamente estudiados; sin embargo, la apoptosis no se circunscribe a células CD4+ infectadas, sino que ocurre también en linfocitos no infectados y en otros tipos celulares, lo cual sugiere la participación de otros procesos en la inmunopatología del VIH, independientes del efecto citopático.2-4El VIH induce una respuesta inmune celular de gran magnitud que aún en presencia de infección progresiva genera células T CD8+ hiperafines.5 Tanto los linfocitos T CD4+ como los CD8+ se activan y proliferan de forma exacerbada durante la infección aguda y crónica por VIH, pero tienen una corta vida media.6 En un principio, el incremento en la tasa de división celular se atribuyó a una respuesta homeostática compensatoria ante la pérdida de células T CD4+.7,8 En pacientes tratados con antirretrovirales se observó que la proliferación de células T caía en forma concomitante con la disminución de la viremia, incluso cuando los linfocitos CD4+ se aproximaban a niveles normales, lo que sugiere que el aumento en la división de células T puede ser un efecto intrínseco del virus.9 La activación inmune crónica es característica de la infección por VIH y conlleva un incremento en la expresión de marcadores de actividad inmunológica que se han utilizado como indicadores de progresión de la enfermedad, tales como: interferón alfa (IFNα), factor de necrosis tumoral alfa (TNFα) y células T CD8+ activadas.10,11
Se considera que el nivel de activación inmunológica es el mejor predictor de enfermedad por VIH, independientemente de la carga viral.12-14 La infección por VIH-2 se caracteriza por una progresión más lenta, menos citopática y con menor viremia y activación inmune que la infección por VIH-1.15,16 Hallazgos documentados en humanos y en primates infectados con virus de inmunodeficiencia del simio (SIV), han cambiado la concepción tradicional de que el efecto citopático viral constituye el desencadenante primario de la depleción de linfocitos T CD4+, hacia la hipótesis de que la activación inmunológica crónica es la causa principal de la depleción celular y la inmunodeficiencia en la infección por VIH.13,17 Muchos estudios destacan la activación inmune como el principal factor de riesgo para la progresión de la enfermedad por VIH, más importante inclusive que la viremia misma e independiente de esta.18
II Causas de activación inmune durante la infección por VIH
El Cuadro 1 resume los mecanismos fundamentalmente implicados en el origen de la activación inmune crónica inducida en el curso de la infección por VIH.
Papel de la inmunidad innata: el compromiso de la inmunidad innata durante la infección aguda por VIH ha sido previamente descrito.17 La infección crónica cursa con elevación de citoquinas proinflamatorias, marcadores séricos de inflamación y activación del sistema de coagulación.19 El ARN monocatenario del VIH estimula los receptores de tipo Toll (toll like receptors, TLR) endosomales 7 y 8.20,21 La unión del ARN viral con TLR7 induce liberación de interferones tipo I (IFN I) por células dendríticas plasmocitoides (pDCs) y constituye un potente activador de células NK (natural killer).22-24 Por otra parte, el reconocimiento intracelular del ADN viral genera una respuesta proinflamatoria con producción de IFNβ e interleukina 1 β (IL-1β).25 El decline abrupto de la replicación del VIH en el inicio de la terapia antirretroviral se asocia con una disminución rápida y progresiva de marcadores de inflamación, adhesinas, moléculas endoteliales, factores de coagulación y activación celular.26
IFN I: los interferones tipo I (IFN I) poseen una importancia crítica en la patogenia de la infección por VIH. Estas citoquinas (principalmente IFNβ y α) se encuentran en la interfase entre la inmunidad innata y adaptativa porque propician la activación y maduración de pDCs, células NK y linfocitos T y B.27 IFNα es el tipo dominante de IFN I detectable en personas infectadas por VIH y sus niveles se relacionan con el grado de depleción de células T CD4+ y activación inmune.28 Polimorfismos en IRF-7 (interferon regulatory factor 7) se han relacionado con el nivel de producción de IFNα en pDCs y con activación de células T CD8+.29 Se ha descrito que pDCs provenientes de mujeres infectadas con VIH producen más IFNα que las de hombres afectados, lo que implica una mayor activación linfocitaria y podría explicar parcialmente la progresión más rápida de la enfermedad en mujeres.30,31
Células supresoras y agotamiento celular: en pacientes infectados con VIH se han identificado diversos tipos de linfocitos T reguladores (Treg), linfocitos B reguladores y células supresoras de origen mieloide. Inclusive ha sido posible detectar Treg antígeno-específicos en los pacientes, lo cual representa un importante avance. Las células supresoras regulan la respuesta inmunológica durante la infección crónica y limitan la inmunopatología asociada con la inflamación, aunque ellas mismas pueden ser también blancos de infección viral. Sin embargo, el bloqueo de las respuestas adaptativas por parte de los linfocitos supresores puede ser perjudicial al prevenir la eliminación del patógeno y favorecer su persistencia, con una exposición antigénica prolongada que conduce al agotamiento de células inmunes (immune exhaustion). Una prioridad es encontrar la mejor estrategia para manipular las células reguladoras que inhiben el establecimiento de una respuesta antiviral eficiente, sin comprometer los mecanismos de contención que evitan el desencadenamiento de un proceso inflamatorio excesivo.32
Cuadro 1 Principales factores involucrados en la fisiopatogenia de la activación inmunológica crónica relacionada con la infección por VIH
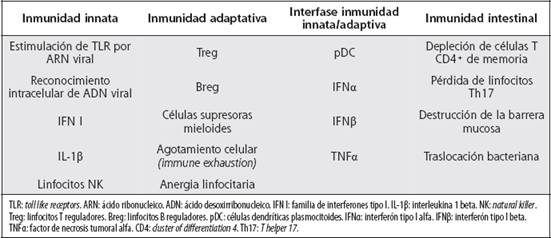
Disrrupción de la homeostasis inmune gastrointestinal: estudios en Macacus rhesus con SIV mostraron una rápida y marcada depleción de las células T CD4+ de memoria del intestino.33 En humanos se describió que estas alteraciones en la inmunidad intestinal resultaban en un incremento del paso de factores bacterianos a la sangre y que la translocación bacteriana provocaba activación inmunitaria sistémica en la infección crónica vía estimulación de TLR en diferentes poblaciones leucocitarias.34-37 Se ha propuesto que la afectación temprana de los linfocitos T CD4+ de memoria en el tracto gastrointestinal puede ser determinante en la progresión de la enfermedad.38 En pacientes que recibían terapia antirretroviral altamente efectiva, el nivel de translocación bacteriana residual se correlacionó con el grado de activación inmunológica y con una falla sostenida en la reconstitución de las células T CD4+, aún en presencia de cargas virales indetectables.39-41La destrucción de la integridad de mucosas en la infección por VIH se asocia con pérdida de linfocitos Th17, los cuales abundan en tejidos mucosos y están vinculados con la inmunidad frente a bacterias comensales.42 Se considera que existe un delicado balance entre Treg y células Th17 que protege contra patógenos, pero evita el daño tisular inducido por una respuesta inmune excesiva.43,44 La dinámica de este equilibrio durante la infección retroviral es motivo de intensa investigación. La pérdida selectiva de linfocitos Th17 intestinales, posiblemente provocada por infección selectiva de esta subpoblación, podría causar disrrupción de la barrera mucosa y activación inmune crónica.42,45
III Efectos patológicos de la activación inmune sobre poblaciones linfocitarias específicas
A continuación se mencionan las consecuencias deletéreas de la activación inmunitaria sobre las principales subpoblaciones linfocíticas (Cuadro 2).
Células T: se acepta el papel protagónico de la activación inmunológica en el desarrollo y progresión de la enfermedad por VIH y su asociación con el descenso de los linfocitos T CD4+. Sin embargo, el resultado final de la infección por VIH no depende solamente de la pérdida de esta población celular, sino también de los efectos adversos de la hiperactivación inmune crónica. La estimulación persistente de la inmunidad adaptativa conduce a la depleción gradual de células T naive con la subsecuente pérdida de la inmunidad específica contra el virus.13,46 Las bases moleculares que determinan los diversos patrones de expansión clonal y contracción en las distintas poblaciones de células T en la infección por VIH permanecen por demás sin dilucidar.47 La inflamación continua en ganglios linfáticos resulta en deposición de colágeno inducido por TGFβ (transformig growth factor β), fibrosis y modificación de la arquitectura ganglionar, lo cual altera la proliferación y supervivencia linfocitarias.48 De manera concomitante, la disfunción tímica y de células progenitoras de estirpe T agrava los efectos inmunodepresores de la enfermedad.49 La activación inmunitaria continua induce sobrerregulación de receptores inhibitorios como CTLA-4 (cytotoxic T-lymphocyte-associated protein 4, CD152), TIM-3 (hepatitis A virus cellular receptor 2, HAVCR2) y PD-1 (programed death-1), que propician anergia e interfieren con la respuesta T antiviral específica.50-52 Asimismo, se ha observado disminución de otras moléculas inmunorreguladoras como BTLA (B- and T-lymphocyte attenuator), lo que podría contribuir a la hiperactivación linfocitaria.53 La activación inmune persistente también tiene efectos deletéreos sobre las células T CD8+, así como en el establecimiento de memoria celular.54-57 La respuesta citotóxica específica posee un papel importante en la inmunidad contra VIH, aunque su relevancia real no se encuentra bien definida.58 Ciertos alelos de HLA clase I se relacionan con la carga viral y la progresión de la enfermedad y pueden ser determinantes en el control viral al afectar la presentación antigénica a linfocitos T CD8+.59
Cuadro 2 Efectos patológicos de la activación inmunológica crónica sobre poblaciones linfocitarias específicas durante la infección por VIH
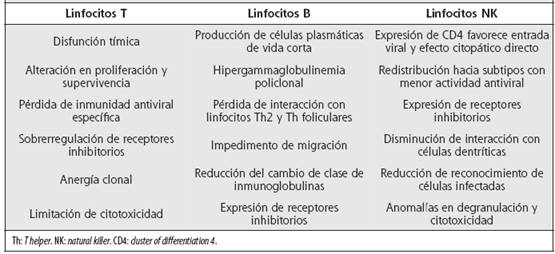
Células B: durante el curso de la infección por VIH, acontece una hiperactivación aberrante de las células B, caracterizada por hipergammaglobulinemia y aumento espontáneo de TNFα e IL-6.60 La reducción de linfocitos B en la infección crónica se asocia con sobrerregulación del receptor PD-1 y disminución de BTLA.61 El aumento en la proliferación y muerte celular, consecuencia de la activación inmunológica crónica por VIH, se relaciona con la estimulación del receptor TLR-9, sobreexpresado en los linfocitos B de memoria durante la infección crónica. Lo anterior conduce a la producción de plasmocitos de vida corta que son responsables de la hipergammaglobulinemia, la cual también entraña mecanismos dependientes de las proteínas virales gp120 y nef.62 El acúmulo de nef en el linfocito B interfiere con la señal del ligando de CD40 (CD40L) e induce un descenso del cambio de clase. Nef también aumenta la expresión de receptores inhibitorios que producen anergia.63 PD-1 altera la interacción de las células B con los linfocitos T ayudadores en el folículo, lo cual disminuye la producción de anticuerpos y la diferenciación en células plasmáticas.64 Se produce además un descenso en la expresión de receptores de quimioquinas, entre ellos CCR7, CXCR4 y CXCR5, necesarios para la migración de células B al nódulo linfático y su interacción con los linfocitos T.65,66 La pérdida progresiva de linfocitos B de memoria compromete la memoria serológica de anticuerpos desarrollados por vacunaciones o infecciones previas.67 Los anticuerpos contra el VIH aparecen semanas después de la infección inicial, pero ciertos tipos de anticuerpos altamente neutralizantes se desarrollan después de varios meses. Dichas inmunoglobulinas específicas podrían ser muy beneficiosas en el control de la infección crónica por VIH, sin embargo,la variabilidad genética del virus y el deterioro en los linfocitos B de memoria afectan la generación de las mismas.68 BTLA es un receptor inhibidor de la actividad inmune que pertenece a la familia CD28. En el linfocito B, su función es inhibida por IFNα secretado por pDC inducidas por el VIH. La inducción de moléculas inhibitorias es parte del proceso de hiperactivación inmune e incide en el progreso de la enfermedad.69
Células NK: en la infección crónica por VIH se induce la expresión de receptores CD4 en los linfocitos NK, lo cual facilita el ingreso del VIH a estas células y favorece sus efectos citopáticos. Además, se produce redistribución de subtipos de células NK hacia poblaciones con menor actividad antiviral (NK anérgicas CD56neg/CD16pos).70 El aumento en la expresión de receptores inhibitorios como PD-1 y la disminución de señales activadoras afectan las funciones de reconocimiento y destrucción de células infectadas que ejercen las células NK.71,72 La carencia de receptores activadores en la superficie de membrana de los linfocitos NK disminuye su interacción con células dendríticas y con ello la producción de citoquinas necesarias para la activación de células NK.73 Determinadas proteínas del VIH pueden alterar las funciones de estos linfocitos. Por ejemplo, vpu inhibe la expresión de ligandos coestimuladores de receptores de células NK en la célula infectada. Nef inhibe la expresión de moléculas MHC clase I (selectivamente HLA-A y HLA-B) y de ligandos celulares (MIC-A, ULBP-1 y ULBP-2) para el receptor activador NKG2D.74 El VIH también altera el mecanismo de degranulación de los linfocitos NK y la citotoxicidad celular dependiente de anticuerpos (antibody dependent cytotoxicity, ADCC), comprometida por disminución del receptor FCγIII.75,76 La citotoxicidad hacia las células infectadas se inhibe y se redirige contra las células CD4+ no infectadas. Este mecanismo se asocia con inducción del ligando NKp44L en la superficie de los linfocitos T CD4+, que estimula la citotoxicidad por parte de de las células NK con una destrucción innecesaria de linfocitos T CD4+ no infectados. NKp44L puede ser estimulado por una fusión abortada del VIH, lo cual se denomina “beso de la muerte”.76,77 En un grupo de pacientes con progresión más lenta de la enfermedad por VIH, se describió la presencia de alelos HLA-C1 y el predominio de genes activadores de receptores KIR, que mejoran la respuesta de los linfocitos NK contra el componente viral Env, lo que se refleja en un mayor descenso de la carga viral.71Polimorfismos de KIR3DS1 y sus respectivos ligandos del grupo HLA-B se relacionan con un curso clínico más lento de la enfermedad.70
IV Consecuencias sistémicas de la inflamación inducidad por VIH y enfermedad no asociada a VIH
La inflamación crónica durante la enfermedad por VIH propicia el establecimiento de un amplio espectro de patologías de origen no infeccioso. Citoquinas proinflamatorias como IL-1, IL-6 y TNFα contribuyen a los efectos patológicos de la hiperactivación inmune suscitada por el VIH.78,79 Inicialmente se atribuyeron condiciones como la aterosclerosis acelerada y la enfermedad cardiovascular al empleo de ciertos antirretrovirales, pero luego se evidenció que el aumento en riesgo cardiovascular es inherente de la población VIH en general, con independencia del tratamiento.80-84 Los portadores de VIH tienen también un riesgo incrementado de desarrollar múltiples complicaciones asociadas con fenómenos de inmunosenescencia no relativos a la infección per se, entre ellas resistencia insulínica, síndrome metabólico, neurodegeneración, alteraciones cognitivas, esteatohepatitis no alcohólica, osteoporosis, insuficiencia renal y enfermedades autoinmunes.19
V Estrategias terapéuticas dirigidas al control de la activación inmunológica
Inmunidad vacunal: las vicisitudes en el desarrollo de vacunas contra el VIH se deben principalmente a la diversidad genética global y la capacidad mutacional del virus, que le permiten evadir la respuesta inmune humoral y celular. El VIH se integra con rapidez en el genoma del hospedero y establece un reservorio latente que no puede eliminarse con los antiretrovirales actuales o las respuestas inmunoespecíficas contra el virus. Hasta el momento no se ha documentado inmunidad natural espontánea con eliminación del VIH que pueda orientar al tipo de respuesta inmune que se debe generar con la vacunación, y solo cuatro conceptos vacunales tienen estudios clínicos de eficacia.85 Diversas vacunas preventivas y terapéuticas contra el VIH han empleado vectores como Listeria monocytogenes, adenovirus, citomegalovirus y lentivirus. Los principales conflictos de seguridad de las vacunas con vectores virales radican en la posibilidad de generar virus patógenos, y en la mutagénesis que puede inducirse en el hospedero al ingresar el ADN del vector.86 Un modelo de vacuna que empleaba adenovirus como vector (ALVAC-HIV) asociado con fragmentos de gag, pol, nef y env, no mostró eficacia en reducir la infección por VIH-1.87 Los vectores de lentivirus han sido muy utilizados en terapia génica, en donde ofrecen la ventaja de replicarse en células en división y en aquellas que no lo están, por lo que son vehículos eficientes para liberar genes de expresión de vida larga. Las vacunas de lentivirus tienen como blanco células presentadoras de antígenos, en las cuales introducen antígenos de VIH que son efectivamente presentados por el MHC. Las células dendríticas diana ofrecen una expresión antigénica más sostenida después de la integración del lentivirus al genoma, mantienen una producción endógena más eficiente de antígenos para la presentación y modulan e incrementan la respuesta de células T. Algunas de las modificaciones efectuadas en el vector lentiviral son la inclusión de grandes cantidades de genes que expresan proteínas inmunoestimuladoras, los cuales fungen como adyuvantes en la respuesta inmune y disminuyen la tolerancia inmunológica generada por las infecciones crónicas. También se ha intentado expresar citoquinas estimuladoras en la célula dendrítica y disminuir moléculas inductoras de apoptosis, lo cual propicia la reactividad y prolonga la longevidad de las células dendríticas que ya fueron diana del lentivirus.86 El empleo de inmunomoduladores como IL-2 con carácter adyuvante puede mejorar la citotoxicidad de las células T CD8+ contra las células infectadas por el VIH.88 No obstante, la adición de IL-2 a la inmunización con ALVAC-HIV, si bien permitió alcanzar un periodo más prolongado de control de replicación viral, no logró evitar nuevos aumentos de la carga viral, no previno el reinicio del TARV y tampoco ejerció una inducción de la inmunidad celular superior a la reportada en portadores de VIH no progresores de larga evolución.89,90 Por su parte, la producción de inmunógenos óptimos que estimulen la inmunidad humoral ofrece muchas dificultades. Para este fin se requeriría, entre otras estrategias, una forma trimérica desdoblada de las glucoproteínas de la cápside viral.91 El diseño de vacunas anti-VIH está limitado por la falta de inmunógenos que generen síntesis de anticuerpos protectores efectivos. La mayoría de los anticuerpos naturales producidos contra las glucoproteínas gp120 y gp41 se dirigen contra zonas con alta tasa de mutación y no generan una respuesta inmune adecuada. Los nuevos modelos vacunales buscan inducir producción de anticuerpos altamente neutralizantes contra gp120 y gp41.92
Control de los efectos adversos de la activación inmune crónica y del daño colateral: se encuentran bajo estudio diferentes inmunomoduladores para disminuir las consecuencias de la activación inmune crónica provocada por VIH. La terapia con IL-2 estimuló la proliferación de linfocitos T CD4+ en pacientes bajo TARV, pero no disminuyó la incidencia de enfermedades oportunistas ni la mortalidad.93 El TARV conduce a un aumento de linfocitos T CD4+, aunque no garantiza una reconstitución completa del repertorio de TCR.94 En este aspecto, la terapia con IL-7 ha demostrado ser una opción segura.95 Esta citoquina estimula la expansión de linfocitos T vírgenes, los cuales exhiben un mayor repertorio de TCR, favorecen la proliferación de linfocitos T de memoria e inhiben la apoptosis de linfocitos T CD4+ y CD8+ en pacientes portadores de VIH-1. Con todo, aún no se ha demostrado la utilidad clínica de estas observaciones.94,96 Algunos estudios pequeños han reportado que la administración subcutánea de hormona de crecimiento induce una mejor reconstitución tímica y de células T CD4+ en pacientes que reciben TARV.97 Investigaciones en primates infectados con SIV, demostraron que la administración de anticuerpos anti-PD-1 estimuló la expansión de linfocitos T CD8+ y linfocitos B de memoria, y además disminuyó la expresión de genes responsables de la transcripción de IFN I.98,99 La ciclosporina se empleó como terapia coadyuvante del TARV en la infección aguda y temprana por VIH, pero no logró establecer un control más temprano de la carga viral.100 La terapia con micofenolato de mofetil no presentó interacciones deletéreas con el TARV, pero su eficacia en el control crónico de los efectos virales no ha sido comprobada.101 Los agentes anti-TNF han mostrado ser seguros en pacientes infectados por VIH que cursan con enfermedad reumática, ya que no inducen variaciones importantes en la cantidad de células T CD4+ ni en la carga viral.102 En primates se ha documentado que el adalimumab se asocia con una menor activación de genes proinflamatorios, sin afectar la carga viral ni la capacidad de activación de los linfocitos T.103 Actualmente se encuentran en desarrollo bloqueadores de TLR-7 y TLR-9. En estudios preclínicos, estas moléculas han mostrado reducir la producción de IFNα en monocitos estimulados por VIH-1.104 Terapias con blanco en Treg VIH-específicos pueden ser beneficiosas si se logra manipular la función de moléculas inhibitorias clave como CTLA-4 y PD-1, estrategia de probada efectividad en cáncer. Otras posibles dianas terapéuticas que han ganado reciente atención son las vías de señalización que involucran CD39 y TIM3-Gal9. Estas nuevas opciones ofrecen la posibilidad de reconocimiento de células supresoras en pacientes infectados, lo cual puede ser útil en el diseño de vacunas terapéuticas. Según las tendencias, se considera que los buenos candidatos vacunales deben no solo generar respuestas efectoras robustas, sino también ser capaces de inhibir vías específicas que conllevan a inmunosupresión y agotamiento inmune.32
En conclusión, la evidencia científica actual involucra al estado inflamatorio persistente asociado con la infección por VIH y causado por múltiples mecanismos, como el principal responsable de la inmunodepleción característica de la enfermedad. La activación inmunológica es inducida por el ARN bicatenario viral y posiblemente por su intermediario de ADN, y en menor instancia por translocación intestinal de productos bacterianos en los pacientes infectados. La muerte linfocitaria secundaria a hiperactivación inmune es adicional a la pérdida celular que resulta del efecto directo del virus. La evidencia parece indicar que la activación inmune crónica es la principal causa de depleción de células T CD4+, la pérdida de inmunidad específica contra el virus y el establecimiento de enfermedades no relacionadas con la infección viral en pacientes que reciben TARV. El conocimiento general de las moléculas implicadas en la inmunopatología del VIH ha aumentado de manera considerable en los últimos años, lo cual ofrece interesantes perspectivas en el desarrollo de nuevas estrategias terapéuticas, incluyendo el diseño de vacunas contra el VIH.
Agradecimientos: a la Dra. María Paz León Bratti (Hospital México, Universidad de Costa Rica), y al Dr. Christian Schauer (Universidad Latinoamericana de Ciencia y Tecnología), por su orientación y apoyo en la realización de este artículo.
Conflictos de interés: los autores declaran que esta revisión fue conducida en ausencia de cualquier relación comercial o financiera que pueda constituir un potencial conflicto de interés.

