











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkLa espondilodiscitis es una condición infecciosa que afecta no solo las vértebras, sino también el disco intervertebral y con frecuencia se aísla Staphylococcus aureus como agente etiológico. El origen de la infección puede ser por inoculación directa durante un procedimiento quirúrgico, o bien, por diseminación hematógena de un foco distante. Cuando hay compromiso del canal vertebral se puede convertir en una emergencia neuroquirúrgica, ya que puede llegar a comprimir la médula espinal y el paciente desarrolla déficit neurológico. Aunque este tipo de infecciones no son raras, hay que destacar algunas condiciones o comorbilidades en las que el paciente está particularmente predispuesto. Tal es el caso de los pacientes portadores de la enfermedad de Rendu-Osler-Weber. Esta entidad, también llamada telangiectasia hemorrágica hereditaria, se asocia con malformaciones arteriovenosas que facilitan la diseminación de una bacteriemia por estafilococo. Los objetivos del artículo son presentar un caso clínico de dicha patología, que demuestra esta asociación en una paciente con severas complicaciones neurológicas y pulmonares, y evidenciar esta predisposición a través de diferentes estudios.
Caso clínico
Paciente femenina de 61 años, con antecedente de tabaquismo crónico por aproximadamente 20 años. Además, conocida portadora de Enfermedad de Rendu-Osler-Weber, e hipotiroidismo desde hace 45 años, en tratamiento de sustitución hormonal con levotiroxina; hipertensa desde hace 15 años en tratamiento con Irbesartán; insuficiencia cardíaca congestiva en tratamiento con furosemida y espirinolactona; enfermedad pulmonar obstructiva crónica (EPOC) desde hace 7 años, en tratamiento con anticolinérgicos y esteroides inhalados. Agrega trastorno de ansiedad en tratamiento con ansiolítico y antidepresivo tricíclico.
Consultó en varias ocasiones al Servicio de Emergencias por cuadro de disnea y epistaxis. Dicho padecimiento fue catalogado como exacerbación del asma bronquial. Pese al tratamiento, la paciente persistió con síntomas, motivo por el cual decidió consultar en clínica privada donde se le diagnosticó un empiema pulmonar, hallazgo puesto en evidencia por un TAC de tórax. Se le realizó drenaje mediante sonda torácica. El análisis bacteriológico del líquido drenado demostró ser un empiema, y de este se logró aislar Staphylococcus aureus multisensible a antibióticos. Se inició tratamiento con vancomicina endovenosa. Eventualmente, al cabo de tres días, la paciente agregó deterioro para la marcha con franca paraparesia espástica y compromiso sensitivo, sin afectación de esfínteres, por lo que se sospechó compresión medular y fue trasladada a otro centro hospitalario para estudiar su nueva patología. En ese lugar se intentó continuar la terapia endovenosa con vancomicina, pero presentó reacción catalogada como alérgica y se cambió el tratamiento a oxacilina endovenosa.
El hemograma mostró hemoglobina en 9,7 gr/dl y el leucograma de 7780/mm3. Las pruebas de coagulación mostraron tiempo de protrombina en 13 segundos (76 % de actividad) y tiempo parcial de tromboplastina en 32,7 segundos. Los marcadores de inflamación reportaron velocidad de eritrosedimentación en 50 mm/hr y proteína C reactiva en 15,8 mg/dl.
Se realizó nueva tomografía de columna torácica, en la cual se demostró severa lesión lítica vertebral a nivel T9 asociado a osteomielitis, compresión medular y posible colección epidural, más colapso del disco intervertebral en T9-T10. La resonancia magnética nuclear confirmó la presencia absceso epidural anterior de la medula espinal (Figura 1).
Se diagnosticó una espondilodiscitis piógena con absceso epidural y se decidió intervenir para drenaje quirúrgico del absceso, toma de cultivos, resección de los cuerpos vertebrales destruidos, lavado y estabilización.
Se realizó una primera intervención de tipo fusión espinal anterior T8 a T10 (6 agosto 2014) con drenaje del absceso y lavado quirúrgico, corpectomía parcial de T 10 y total de T9, con colocación de malla intersomática con injerto autólogo, disquectomía T8-T9 y T9-T10 y colocación de sonda de tórax. Luego se efectuó una segunda intervención de estabilización, abordaje posterior con instrumentación de T7-T12 con tornillos transpediculares. Los cultivos del material discal y vertebral resecado fueron negativos, producto de la antibioticoterapia que recibió durante 2 semanas.
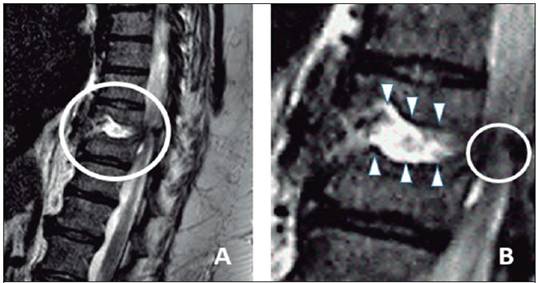
Figura 1: Resonancia magnética nuclear de columna presenta lesión lítica vertebral a nivel de T 9 (A). Se muestra en el acercamiento (B) colapso del espacio entre T9-T10 (puntas de flecha) y asocia compresión medular más colección epidural (círculo).
En el postoperatorio inmediato la paciente requirió ventilación mecánica por alteración del sensorio y patrón respiratorio inadecuado. Agregó posteriormente neumonía asociada al ventilador con secreción bronquial purulenta y elevación de marcadores inflamatorios, por lo cual se hizo cultivo de secreción bronquial.
Se dio cobertura antibiótica contra gérmenes gram negativos y luego se confirmó aislamiento bacteriológico positivo por Pseudomona aeruginosa multisensible (se trató con amikacina, ceftazidime, oxacilina y levofloxacina). Dado que fue difícil el proceso de desacostumbramiento ventilatorio, se decidió en el postoperatorio 9, realizar traqueostomía. Se logró independizar del ventilador mecánico, y se inició movilización al día 5. Se consiguió egresar de la Unidad de Cuidado Crítico. Continuó proceso de rehabilitación y completó 4 semanas de tratamiento antibiótico. Posteriormente se egresó con tratamiento antibiótico oral por 2 semanas más, con adecuada evolución general.
Discusión
La telangiectasia hemorrágica hereditaria es una enfermedad autosómica dominante con una incidencia de 1 caso por 5000 a 8000 habitantes.1 Fue descrita por primera vez por Henry Gawen Sutton, en 1864.2 Henri Jules Louis Marie Rendu describió, en 1896, un caso en un paciente con epistaxis recurrente.3 William Osler estableció su origen hereditario y Frederich Weber realizó la descripción clínica en una serie de casos.4,5 La entidad se caracteriza por la presencia de epistaxis recurrente, telangiectasias cutáneas y malformaciones arteriovenosas viscerales que afectan los pulmones, el tracto gastrointestinal y el cerebro.6
Para el diagnóstico se utilizan los criterios de Curazao, considerándose definitivo si se cumplen por lo menos 3 de los siguientes hallazgos: epistaxis espontáneas recurrentes, presencia de telangiectasias, historia familiar de la condición y presencia de lesiones viscerales.7
Desde el punto de vista genético hay alteraciones en la endoglina (THH tipo I), kinasa similar al receptor activina (THH tipo II) y la proteína Smad 4 (THH tipo II), asociándose el tipo 4 con alteraciones del cromosoma 5 y el tipo IV a al cromosoma 7.8
Uno de los hallazgos más frecuentes es la presencia de malformaciones arteriovenosas en cerebro, hígado y, muy especialmente, a nivel pulmonar, lo que se presenta entre el 15 al 33% de los pacientes, los cuales se complican con hipertensión pulmonar.9 Las malformaciones en este nivel pueden asociarse a embolias sépticas y a la producción de abscesos cerebrales.10 En el presente caso no se realizaron estudios adicionales para descartar malformaciones arteriovenosas en algún sitio.
La predisposición a infecciones en este tipo de pacientes incluye sitios cerebrales y extracerebrales.11,12 Por su parte, Dupuis et al. reportaron 4 casos de infección espinal, que corresponden a un 6,2 %.11 Entre los sitios extracerebrales se han descrito: endocarditis bacteriana, empiema, abscesos hepáticos y espondilodiscitis, siendo frecuentemente aislado Staphylococcus aureus como germen responsable.13,14 Musso y colaboradores lograron identificar en su experiencia, únicamente 1 caso de espondilodiscitis,13 debido a lo cual, el caso que se presenta puede considerarse particularmente inusual.
El mecanismo para dicha predisposición podría estar vinculado con la ocurrencia de episodios de epistaxis, lo cual conlleva, principalmente por uso de taponamiento, a colonización por este germen.11,13,15 A diferencia de los abscesos cerebrales, las infecciones extracerebrales podrían estar más bien asociadas a esta colonización y no obedecer a las malformaciones arteriovenosas pulmonares. Aunque se ha estudiado poco, también se ha logrado determinar que hay deficiencia en la capacidad fagocítica de neutrófilos y monocitos en estos pacientes.16 No obstante, Blanco y colaboradores no lograron encontrar disfunción linfocitaria en esta población.
El caso reportado de la paciente con síndrome Rendu Osler Weber, ilustra la presencia de un proceso infeccioso por estafilococo, lo cual obliga a descartar sitios extracerebrales tipo espondilodiscitis, como foco infeccioso oculto. Es de especial interés que la presentación inicial fue a nivel respiratorio, lo que desvió la causa real del proceso infeccioso encontrado luego a nivel vertebral, de manera que es necesario sospechar este tipo de afecciones en presencia de algún proceso infeccioso de foco no identificado.

