










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Costarricense
versión On-line ISSN 0001-6002versión impresa ISSN 0001-6012
Acta méd. costarric vol.50 no.1 San José mar. 2008
Actualización en los mecanismos fisiopatológicos de la insuficiencia cardiaca
(Update in physiopathological mechanisms in heart failure)
Guido Ulate-Montero,Adriana Ulate-Campos
Resumen
La insuficiencia cardiaca es uno de los síndromes clínicos más frecuentes en la práctica médica y se presenta cuando el corazón,a presiones normales de llenado,es incapaz de bombear la cantidad suficiente de sangre que requiere el metabolismo de los tejidos.Aquí se presenta una revisión de los datos más recientes sobre los mecanismos involucrados en la fisiopatología de la insuficiencia cardiaca,para que su conocimiento sea de utilidad en el manejo adecuado de esta entidad. En la insuficiencia cardiaca,como respuesta al gasto cardiaco insuficiente, se activan una serie de mecanismos neuroendocrinos sistémicos,que posteriormente, contribuyen al deterioro del cuadro clínico; es el caso del sistema simpático y el sistema renina-angiotensina-aldosterona, los cuales terminan produciendo daño endotelial, incremento de radicales libres, de la apoptosis, de la fibrosis cardiaca y generación de arritmias.También se observa un incremento en la liberación de péptidos natriuréticos, los cuales tienden a regular algunas de las respuestas neurohumorales exacerbadas, sin embargo, con el transcurso de la enfermedad su acción tiende a atenuarse.Celular y molecularmente se producen una serie de alteraciones en el manejo intracelular del Ca2+, así como en algunas de las corrientes iónicas que participan en la generación del potencial de acción de los miocitos cardiacos. La remodelación cardiaca precede al cuadro clínico de la insuficiencia y contribuye a su deterioro.Mensajeros químicos como la endotelina-1, la norepinefrina y la angiotensina II, que activan la cascada de las MAP quinasas, provocan hipertrofia cardiaca, lo que favorece la isquemia y la aparición de arritmias.El manejo farmacológico de la insuficiencia cardiaca debe dirigirse a los mecanismos fisiopatológicos afectados, es decir, al bloqueo de las acciones deletéreas de los sistemas neuroendocrinos sobreestimulados y a evitar la pérdida de miocitos, la generación de fibrosis y de arritmias cardiacas, para lo cual es indispensable el manejo apropiado de los niveles intracelulares de Ca2+.
Descriptores: Insuficiencia cardiaca, remodelación cardiaca, activación neurohumoral, acople excitación-contracción, arritmias, apoptosis.
Abstract
Heart failure is one of the most frequent clinical syndromes in medical practice;it appears when the heart is unable to pump enough volume of blood to supply the tissue s requirements.This article reviews the most recent information regarding the mechanisms involved in the pathophysiology of heart failure,the main goal is to offer the knowledge necessary to understand and manage properly this condition.In heart failure,as a response to the low cardiac output,a series of neuroendocrine systemic mechanisms are activated,but they contribute to deteriorate the clinical status;this happens with the sympathetic and the renin-angiotensin-aldosterone systems,which end up producing endothelial damage,increase of oxidative radicals,apoptosis, cardiac fibrosis and generation of arrhythmias.Also there is an increase in the secretion of natriuretic peptides,which tend to regulate some of the exacerbated neuroendocrine responses, but with time their effect tend to diminish.At the cellular and molecular level a series of alterations occur in the regulation of intracellular Ca2+, as well as in some of the ionic currents that play a role in the generation of action potentials in cardiac myocytes.Cardiac remodeling precedes the clinical manifestations of heart failure and contributes to its deterioration.Chemical messengers like endothelin-1,norepinephrine and angiotensin II,activate the MAP kinases cascade and provoke cardiac hypertrophy favoring the development of ischemia and the appearance of arrhythmias.Pharmacological management of heart failure must aim the mechanisms affected,it must block the deleterious actions of the neuroendocrine systems,avoiding the loss of myocytes, the generation of fibrosis and the production of cardiac arrhythmias,in order to achieve this goal an appropriate management of intracellular levels of Ca2+ is required.
Key words:Heart failure,cardiac remodeling,neurohumoral activation,excitation-contraction coupling, arrhythmias,apoptosis
Recibido: 31 de julio de 2007 Aceptado: 5 de septiembre de 2007
Después de 100 años de que Ringer descubrió que el Ca2+ era esencial para la contracción normal del corazón, se ha comprendido que la modulación intracelular de la concentración de Ca2+, por las proteínas del retículo sarcoplásmico, constituye una promesa para luchar contra la insuficiencia cardiaca.1
La insuficiencia cardiaca (IC)es un síndrome clínico de pronóstico pobre, caracterizado por un conjunto de signos y síntomas que se presentan tanto como resultado de un gasto cardiaco (GC) insuficiente,como de una serie de mecanismos compensadores cardiovasculares y neurohumorales.
Clásicamente,se ha dicho que existe IC cuando el corazón, a presiones normales de llenado,es incapaz de bombear la cantidad suficiente de sangre que requiere el metabolismo de los tejidos.2 Puede ocurrir en solo uno de los ventrículos o en ambos, y deberse a problemas en la eyección (IC sistólica)o en el llenado (IC diastólica).3 El resultado final es una serie de eventos complejos que provocan un conjunto de respuestas neuroendocrinas,4-9 las cuales conducen al "círculo vicioso "que caracteriza la evolución de la IC y que será ampliamente tratado en esta revisión.
En el mundo,más de 20 millones de personas padecen de IC8,10 y a pesar de los adelantos en el manejo de esta enfermedad, la mayoría de los pacientes con las formas avanzadas mueren un año después de haber sido diagnosticados.11
Estudios de poblaciones,como el de Framinghan,12 que le ha dado seguimiento por 34 años a 5 209 personas, registra prevalencias de IC del 0.8%,el 2.3%,el 4.9%y el 9.1%, en los grupos etáreos de 50-59,60-69,70-79 y más de 80 años, respectivamente.
El paradigma utilizado en la explicación de la fisiopatología de la IC ha pasado por diversos enfoques y no está totalmente dilucidado. Hoy, este síndrome debe analizarse desde un punto de vista sistémico que incluye la activación neurohumoral propia de esta condición, acompañado de los cambios que ocurren celular y molecularmente y explican la remodelación cardiaca, la apoptosis y la propensión a padecer arritmias.
El objetivo de esta revisión es brindar al médico un conocimiento actualizado sobre los mecanismos que explican los hallazgos clínicos de la IC y su evolución, para que él pueda ofrecer a los pacientes que padecen este síndrome, un tratamiento cada vez más eficiente, en el cual se vean integrados dichos conocimientos. Sin embargo, siempre debe enfatizarse la importancia de adoptar las medidas preventivas en contra de todos aquellos factores de riesgo que conducen a la IC.
Cuadro clínico
El cuadro clínico de la IC es complejo y aún no está totalmente comprendido.4 Frecuentemente se presentan síntomas y signos por intolerancia a los esfuerzos, como la disnea y la fatiga, y otros por retención de líquidos como el edema periférico y la congestión pulmonar.13
Aunque la IC se considera un desorden hemodinámico, varias investigaciones han mostrado una pobre correlación entre las mediciones de la funcionalidad cardiaca y los síntomas que presentan los pacientes.3,14
Clínicamente,la IC se caracteriza por la presencia de edema periférico, disnea, fatiga, ortopnea, disnea paroxística nocturna, cardiomegalia, y un tercer ruido en la auscultación.15
Se ha utilizado el término de IC congestiva (ICC)cuando el acúmulo de líquido y la retención de sodio producen una congestión severa que causa un incremento de la presión venosa yugular,cambios pulmonares,edema periférico o hepatomegalia.4
Etiología
En el mundo occidental, las 3 principales causas de IC son: la cardiopatía hipertensiva,la cardiopatía isquémica asociada con un infarto previo y la miocardiopatía dilatada.3,4,15 Otras causas son: arritmias,valvulopatías, infecciones, enfermedades por infiltración, alcoholismo, endocrinopatías y enfermedades genéticas.
Disfunción sistólica o diastólica
Clínicamente, la IC puede presentarse por disfunción sistólica o diastólica. En la IC sistólica la alteración ocurre por decremento en el número de miocitos o por disminución en la función contráctil de estos,lo que afecta la eyección.17,18 En la IC diastólica el problema radica primariamente en una pobre relajación del miocardio, que afecta el llenado ventricular.4,18 Hemodinámicamente, en la primera existe clara depresión de la fracción de eyección (<40%);en la segunda, este parámetro está menos comprometido (>40%).
Los pacientes con IC secundaria a cardiopatía hipertensiva generalmente inician con sintomatología por disfunción diastólica, es decir, disnea de esfuerzo, y tienden a presentar un ventrículo izquierdo (VI) con hipertrofia concéntrica.Los pacientes con IC secundaria a isquemia o
a una miocardiopatía idiopática, desarrollan IC por disfunción sistólica y usualmente presentan un VI dilatado.19 Jessup y Brozena presentan un cuadro con las principales diferencias entre estos 2 tipos de disfunciones, sin embargo, se debe aclarar que en general, y sobre todo después de cierto tiempo de evolución, ambas se presentan concomitantemente, por lo que sería más adecuado hablar en términos de la disfunción predominante.4
Mecanismos fisiopatológicos
Clásicamente, la fisiopatología de la IC ha sido estudiada como una serie de mecanismos reflejos, tales como la activación adrenérgica y la del sistema renina-angiotensina-aldosterona, que se desencadenan ante una disminución de la presión arterial y que promueven un incremento en las
resistencias periféricas y en la poscarga cardiaca.4 Al inicio, la retención de sal y líquido funcionan, de acuerdo con el "mecanismo de Frank-Starling", incrementando el GC, pero luego se presenta la congestión y el cuadro empeora.20
Las afirmaciones del párrafo anterior siguen siendo válidas y corresponden a un resumen muy general de la fisiopatología de la IC. Actualmente, se conoce mucho más del tema. A continuación se profundiza en aquellos mecanismos cuyo conocimiento es indispensable para comprender la evolución y la respuesta al tratamiento de los pacientes que padecen IC.
Papel del calcio en los miocitos cardiacos
La evolución de la IC se asocia con cambios en los mecanismos moleculares que explican el acople excitación-contracción. Para entenderlos, se revisará, de manera sencilla, el funcionamiento normal de estos mecanismos.
En los miocitos cardiacos,la contracción es disparada por el Ca2+ que ingresa desde el líquido extracelular (LEC) a través de canales iónicos tipo "L", presentes en el sarcolema y cuya apertura es dependiente de voltaje, es decir, se abren durante la fase 2 ó "de meseta" del potencial de acción (PA), cuando la membrana está despolarizada.17,21 El Ca2+ que ingresa provoca la liberación de más Ca2+ a partir de las reservas existentes en el retículo sarcoplásmico (RS), mecanismo conocido como "liberación de Ca2+ inducida por Ca2+". 1,21,22 La salida de estas reservas ocurre a través de los llamados receptores de rianodina (RyR2),que son proteínas en la membrana del RS que también funcionan como canales para Ca2+.10,21 Una vez que el Ca2+ aumenta en el citosol, interactúa con los miofilamentos y provoca la activación de los puentes cruzados, y con ello la contracción.
La relajación del músculo cardiaco (lusiotropismo)está mediada por la recaptura del Ca2+ al RS, así como por la expulsión del ión hacia el LEC. Una ATPasa, conocida como la SERCA2a, es la proteína transportadora encargada de realizar esta recaptura en contra del gradiente de concentración. Las proteínas NCX1 y una Ca2+ ATPasa del sarcolema expulsan el Ca2+ hacia el LEC.21
La actividad de los RyR2 es finamente modulada por múltiples reguladores; uno de ellos, la calstabina2, también conocida como FKBP12.6, merece especial interés, pues durante la diástole, se une a los RyR2 y los mantiene cerrados. En la IC, cuando los RyR2 son fosforilados por la proteína quinasa A (PKA), como resultado de la sobreestimulación adrenérgica, la calstabina se separa de los RyR2 y el Ca2+ sale del RS, en mayor proporción. Esta salida aumentada tiene 2 consecuencias: (1) la generación de postdespolarizaciones tardías que pueden disparar una taquicardia ventricular y provocar la muerte súbita,10 y (2) la disminución de las reservas de Ca2+ del RS17, debido a que aumenta la proporción de este ión que es expulsado hacia el LEC y disminuye la que es recapturada en el RS. Lo anterior ocurre debido a que los transportadores NCX1 se encuentran muy activos y, en cambio, las SERCA2a están deprimidas.
La actividad de la SERCA2a es regulada por el fosfolamban: una proteína ubicada en la membrana del RS, que al interactuar con ella la inhibe, pero cuando el fosfolamban está fosforilado, dicha inhibición pierde importancia.1,22 En la IC se reduce la fosforilación del fosfolamban, lo que contribuye, como ya fue explicado, a la disminución de la recaptura del Ca2+ en el RS.17,23,24
El Ca2+, además de cumplir una función en la contracción y relajación de los miocitos, también es importante como segundo mensajero en diferentes vías de señalización, dentro de las cuales están aquellas que, en la IC, se relacionan con la remodelación cardiaca.
Remodelación cardiaca
Conforme la IC progresa se producen cambios en la morfología y la estructura del ventrículo izquierdo, conjunto conocido como remodelación cardiaca.
Generalmete, el inicio de la remodelación cardiaca precede el desarrollo de los síntomas, pero continúa después de la aparición de estos y contribuye sustancialmente en el deterioro del cuadro clínico. Ciertos mensajeros químicos que se activan durante el desarrollo de la enfermedad, como la angiotensina II (ANGII), la aldosterona, la endotelina, la vasopresina (HAD) y las citoquinas, son los responsables de esta remodelación.3
Los miocitos cardiacos adultos son células diferenciadas que han perdido la capacidad de división, por lo tanto, un aumento en la masa cardiaca está básicamente determinado por un crecimiento celular. El problema que se presenta durante este crecimiento es que también ocurren alteraciones específicas en la expresión genética y en su fenotipo, algunas de tipo adaptativo, como ocurre con la expresión de genes que codifican para péptidos natriuréticos y para proteínas contráctiles fetales, y otras de carácter no adaptativo, como sucede con la fibrosis.27
Los cambios en el tamaño de la cavidad y en la estructura, además de incrementar la tensión sobre las paredes del corazón insuficiente, también deprimen su desempeño mecánico y pueden aumentar la regurgitación a través de la válvula mitral.3,28
Cuando la hipertrofia es secundaria a una sobrecarga de volumen (insuficiencia aórtica o mitral), el crecimiento de los miocitos ocurre por adición de nuevos sarcómeros en serie (uno detrás del otro); como resultado de ello, los miocitos se alargan y el corazón presenta una hipertrofia excéntrica (dilatada), es decir, el radio de la cavidad crece. En cambio, cuando la hipertrofia ocurre secundaria a una sobrecarga de presión (hipertensión arterial o estenosis aórtica), los sarcómeros se agregan en paralelo (uno a la par del otro), lo que provoca un engrosamiento de la pared ventricular, es decir, una hipertrofia concéntrica, en la cual el radio de la cavidad disminuye. De acuerdo con la ley de Laplace, la tensión en la pared de un ventrículo que sufre una hipertrofia excéntrica es superior a la que sufre la pared de un ventrículo con hipertrofia concéntrica, debido a que la tensión es directamente proporcional al radio de la cavidad e inversamente al grosor de la pared.
La reserva de irrigación coronaria se encuentra disminuida cuando hay hipertrofia del VI, lo que se traduce en un mayor riesgo de isquemia durante el ejercicio, especialmente en aquellas regiones endocárdicas sujetas a mayor tensión.4 Las crisis de isquemia eventualmente pueden conducir a la muerte de los miocitos.
Aunque al inicio el papel de la hipertrofia es compensatorio, posteriormente lleva al deterioro de la enfermedad, conduciendo a la IC congestiva y al aumento en la frecuencia de la producción de arritmias.29 El mecanismo por medio del cual ocurre esta transición no está claro.Se ha postulado que ciertos mensajeros químicos como la endotelina-1 (ET-1), la norepinefrina (NE),7 la ANGII30 y algunos factores de crecimiento, todos los cuales se encuentran aumentados en la IC, terminan activando ciertas fosfolipasas que luego activan a la proteinquinasa C (PKC), con lo que provocan, por medio de la cascada de las MAP quinasas,un aumento en la síntesis proteica y, con ello, la hipertrofia.25,31 Por otro lado, los cambios que ocurren en la concentración de fosfolípidos en el sarcolema de los miocitos de corazones insuficientes terminan produciendo modificaciones en la actividad de algunos transportadores, como por ejemplo:en el de sodio-calcio (NCX1), que aumenta su actividad; en canales iónicos como los rectificadores de entrada de potasio, que también incrementan su probabilidad de apertura, así como en las conexinas expresadas por los miocitos.29 Todos estos cambios favorecen la aparición de arritmias cardiacas.
Activación neurohumoral
En la IC existe alteración neuroendocrina en la que, el sistema simpático, el sistema renina-angiotensina-aldosterona (SRAA), la vasopresina (HAD), y los péptidos natriuréticos, entre otros, desempeñan un papel preponderante.7,8,32-34 En este sentido, la integridad de la circulación arterial se debe analizar como el balance que normalmente tiene que existir entre el gasto cardiaco (GC) y las resistencias arteriales periféricas (RP).9 En la IC se presenta un desbalance entre estos 2 parámetros, que conduce a la activación de los sistemas mencionados.
El sistema adrenérgico
Inicialmente, la disminución de la función miocárdica conduce a un aumento en la actividad del sistema simpático, que se traduce en un incremento en la frecuencia y la contractilidad cardiaca, lo que compensa parcialmente el problema. Sin embargo, conforme la IC avanza, la actividad simpática aumenta aún más, pero, con el paso del tiempo, esto conlleva a un deterioro en la estructura y la función miocárdica;8,35-37 que ha sido asociado principalmente, con la activación de la vía de señalización correspondiente a los receptores ß1 adrenérgicos.38-40
La cascada de señalización desencadenada por la activación de los receptores ß1 adrenérgicos activa la proteinquinasa dependiente de cAMP (PKA). Esta quinasa, al fosforilar ciertas proteínas, modifica en funcionamiento. Algunas de ellas son: 1-los canales de calcio tipo "L" del sarcolema y los RyR2,que al ser fosforilados por la PKA aumenta la probabilidad de su apertura,41,42 2-el fosfolamban que al ser fosforilado disminuye su acción inhibitoria sobre la SERCA2a,43 3-miofilamentos como: la troponina I y la proteína C que une a la miosina (MyBP-C) y al ser fosforiladas reducen su sensibilidad al Ca2,44-46 y 4-los mismos receptores ß adrenérgicos que al ser fosforilados sufren desensibilización.8,47-49
La desensibilización de los receptores ß adrenérgicos conduce a una menor activación de la vía de señalización adrenérgica, incluyendo una menor fosforilación de aquellas proteínas que son blanco de la PKA. Sin embargo, se debe aclarar que en los miocitos de los corazones insuficientes, debido a la presencia de otras quinasas o a la disminución de la actividad de las fosfatasas, no siempre se presenta una disminución en las fosforilaciones. Por ejemplo, se ha observado que tanto los canales tipo "L" como los RyR2 están hiperfosforilados, a pesar de la desensibilizción.50-53 El fosfolamban, en cambio, se encuentra menos fosforilado, lo que hace que la actividad de la SERCA2a sea menor, así como la recaptura del Ca2+ hacia el RS, produciéndose, como ya fue dicho,una depleción del calcio almacenado y un efecto inotrópico negativo.8,54,55
También se ha demostrado que la exposición prolongada de los miocitos cardiacos a altas concentraciones de catecolaminas, tiene un efecto tóxico que conduce a la degradación de las miofibrillas y al aumento del colágeno, lo que se traduce en cambios inotrópicos y lusiotrópicos negativos.56,57
El sistema renina-angiotensina-aldosterona
En la IC, tanto la caída en la perfusión renal como la activación del sistema adrenérgico,estimulan la liberación de renina y con ello también la de la ANGII y la de la aldosterona. Estos 2 últimos mensajeros químicos poseen una serie de efectos, compensadores algunos y deletéreos otros, que influyen sobre la evolución de la IC. La ANGII, además de producir, directamente, una vasoconstricción periférica, también estimula la activación 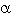 -adrenérgica y la liberación de aldosterona. Dentro de los efectos adversos están: 1-daño del endotelio vascular, 2-incremento de radicales oxidativos intracelulares,4 y 3-efecto mitógeno en miocitos cardiacos.9
-adrenérgica y la liberación de aldosterona. Dentro de los efectos adversos están: 1-daño del endotelio vascular, 2-incremento de radicales oxidativos intracelulares,4 y 3-efecto mitógeno en miocitos cardiacos.9
El nivel en que aumenta la actividad de renina y la concentración de aldosterona en el plasma de los pacientes que padecen de IC, guarda relación con la severidad y el pronóstico de esta patología.32,58 Además, se sabe que el fenómeno de escape a la acción de los niveles altos y sostenidos de aldosterona no ocurre en estos pacientes,59 lo que permite la retención continua de sodio y el empeoramiento de la congestión. Aunque no está del todo claro el motivo por el cual no hay escape, se piensa que una disminución de la carga de sodio que se presenta al túbulo colector, debido a un aumento en la reabsorción del túbulo proximal por efecto -adrenérgico y de la misma ANGII, sería parte de la explicación.9
Por otro lado,recientemente se han demostrado una serie de efectos directos de la aldosterona que quizás también inciden en la evolución de la IC, como: (1) estrés oxidativo y disminución en la producción de óxido nítrico,60 (2) eventos inflamatorios vasculares,61 (3)fibrosis cardiaca,61 (4) taquicardia 2 y (5) generación de arritmias, que aunque no está claro el mecanismo, se ha asociado con modificaciones en la corriente de Ca2+ a través de los canales "L" y "T" presentes en el sarcolema.61,63
Liberación no osmótica de la hormona antidiurética
En algunos pacientes con IC se puede presentar una retención de agua superior a la de sodio, lo que conduce a hiponatremia. De hecho,este hallazgo es un signo de mal pronóstico.
La liberación de HAD ha sido implicada en esta retención, y se presenta aún cuando la osmolalidad plasmática está baja.9 Pareciera entonces que la disminución de la presión en los receptores del seno carotídeo sea el mecanismo responsable.4
Péptidos natriuréticos
En la IC se observa un incremento en los niveles sanguíneos del péptido atrial natriurético (PAN)y del péptido cerebral natriurético (PCN).65,66 Ambos péptidos son producidos y secretados por el corazón ante un aumento de la distensión de las cavidades cardiacas; el PAN por los miocitos atriales y el PCN por los miocitos ventriculares. El primero parece secretarse principalmente cuando se producen cambios agudos, y el segundo, ante los cambios crónicos.61
Los péptidos natriuréticos poseen varias acciones:1- tienden a inhibir al sistema simpático y al SRAA, 2-facilitan la natriuresis y la diuresis, 3-disminuyen las resistencias periféricas, 4-relajan el músculo liso y 5- tienden a contrarrestar la acción de los mensajeros químicos que producen la remodelación cardiaca.61
En un principio, todas las acciones de los péptidos natriuréticos estarían fisiológicamente justificadas, pues tienden a compensar los efectos deletéreos ocasionados por la activación de sistemas como el simpático y el SRAA. De hecho, si bien es cierto que muchos de los mecanismos compensadores presentados en esta revisión terminan deteriorando aún más la función cardiaca, este no sería el caso de los péptidos natriuréticos, los cuales participan disminuyendo tanto la postcarga como la precarga, que están elevadas debido al círculo vicioso en que se entra por la activación neurohumoral.4 El problema, sin embargo, es que con el transcurso de la enfermedad tiende a atenuarse su liberación y a presentarse una regulación a la baja de sus receptores.67,68
Apoptosis
Cuando se comparan los corazones insuficientes con los sanos, en los primeros se observa un mayor número de miocitos que presentan apoptosis:aproximadamente de 25 a 80 veces más.69-71
La apoptosis es un proceso de suicidio celular que, en términos evolutivos, ha sido importante en el desarrollo embrionario y postnatalmente lo será en la remodelación, la homeostasis, la sobrevida celular y los procesos inmunológicos.72
Cuando los mecanismos productores de apoptosis pierden su regulación, aparece un desbalance en la homeostasis y se generan ciertas enfermedades; este es el caso de la IC. Los miocitos sufren apoptosis en respuesta a estímulos como: la hipoxia, la acidosis, el estrés oxidativo, la ANGII,los agonistas ß adrenérgicos, el estiramiento, la hipoglicemia y el factor á de necrosis tumoral. Casi todos estos estímulos están presentes en la IC.69,72,73
Los miocitos apoptóticos son reemplazados por tejido fibrótico, lo que compromete la capacidad contráctil y aumenta la probabilidad de generación de arritmias.
La estimulación adrenérgica del tipo ß 1 tiende a aumentar la apoptosis de los miocitos cardiacos,39 mientras que la del tipo ß2 tiende a inhibirla.4,74-76
La apoptosis es mediada por caspasas: enzimas proteolíticas que hidrolizan las uniones peptídicas del grupo carboxilo de los residuos de ácido aspártico. Existen evidencias que demuestran que en algunos modelos de IC la inhibición de las caspasas disminuye la apoptosis y mejora el pronóstico,72,77,78 lo que parece reafirmar la importancia de este proceso de suicidio celular en la evolución de la IC.
Alteraciones electrofisiológicas
La mitad de las muertes por ICC son causadas por taquicardia ventricular y cerca del 80%de los pacientes con IC sistólica presentan este tipo de arritmia.10
La duración del PA en los miocitos de corazones en IC está prolongada, debido principalmente, a una reducción en la corriente de K+ durante la fase 1.17,79
Por otro lado, las corrientes de calcio a través de los canales tipo "L" y tipo "T" se ven alteradas, lo que se ha asociado al hiperaldosteronismo que se presenta en la IC.61 Estos cambios en las corrientes iónicas de los miocitos serían parcialmente responsables de una mayor probabilidad de generación de arritmias.
Otro mecanismo productor de arritmias cardiacas y de muerte súbita es, como ya se dijo, la activación de los RyR2, que permite una mayor salida de Ca2+ del RS, y favorece la generación de postdespolarizaciones tardías que pueden disparar una taquicardia ventricular.1 De hecho, recientemente se ha demostrado que un medicamento llamado JTV519, que es una benzotiacepina,estabiliza el estado cerrado de los RyR2 previniendo la aparición de arritmias.80
Por último,debe aclararse que en la IC,la posibilidad de la aparición de arritmias cardiacas también posee un componente genético.81
Farmacodinamia de algunos de los medicamentos utilizados en el manejo de la IC
El objetivo de esta revisión no es presentar el manejo detallado de la IC, pero sí la información necesaria para comprender los mecanismos farmacodinámicos y las razones por las cuales se utilizan determinados agentes terapéuticos.
El manejo de la IC clásicamente ha estado dirigido a: disminuir la presión venosa central con diuréticos, reducir la postcarga con vasodilatadores periféricos y aumentar la contractilidad cardiaca con agentes inotrópicos. Desafortunadamente, los estudios clínicos realizados con
estos fármacos han arrojado resultados desalentadores, ya que se ha logrado poco en lo que respecta a la prolongación de la sobrevida de estos pacientes.8
Con respecto a los bloqueadores beta adrenérgicos, se ha reportado que después de 3-4 meses de su utilización, mejora la función sistólica y revierte la remodelación, lo cual se explica por el control que ellos producen sobre la estimulación simpática exacerbada en la IC crónica.13,82,83 Sus efectos se asocian con un aumento en la sobrevida y en la fracción de eyección,una disminución en el número de hospitalizaciones y en la incidencia de muerte súbita.
El uso de los inhibidores de la ECA y los bloqueadores de los receptores para ANGII ha mejorado la sobrevida de los pacientes con IC.Se ha visto que este tipo de fármacos, además de disminuir la poscarga, reducen la hipertrofia ventricular izquierda.9 Los diuréticos, especialmente las tiazidas y los de asa, mantienen su lugar preponderante, pues permiten controlar la sobrecarga de volumen y los síntomas propios de la congestión. La espironolactona, así como otros inhibidores de los receptores de aldosterona que están en estudio, evitan la retención de sal, la pérdida urinaria de potasio y, lo más novedoso, disminuyen la fibrosis cardiaca.84
Los digitálicos,además de su clásico efecto inotrópico positivo,aumentan la descarga de las aferencias de los receptores de presión cardiacos, lo que se acompaña de una disminución de la actividad simpática sobre el corazón.7 Sin embargo, su utilización se ha asociado con un mayor riesgo de presentar arritmias.13
El manejo farmacológico de la IC ha sido básicamente paliativo y poco dirigido a los mecanismos moleculares afectados.1 En este sentido, nuevos tratamientos, muchos de ellos en investigación, se enfocan a la reversión de algunos de los mecanismos alterados, como, la disminución en la actividad de las SERCA2a o la inestabilidad de los RyR2. Un ejemplo es el JTV519,un derivado de las benzotiacepinas que favorece la unión de la calstabina2 a los RyR2 y así previene las arritmias y la muerte súbita.1,10 También se realizan investigaciones con fármacos que evitan la apoptosis,que sensibilizan los miofilamentos al Ca2+ y que bloquean los receptores V2 para ADH.85
Referencias
1. Minamisawa S, Sato Y, Cho MC. Calcium cycling proteins in heart failure, cardiomyopathy and arrythmias.Exp Mol Med 2004;36:193-203. [ Links ]
2. Braunwald E, Colucci W, Grossman W. Aspectos clínicos de la insuficiencia cardíaca: insuficiencia cardíaca de gasto alto; edema pulmonar. En: Zipes DP, Libby P, Bonow RO, Braunwald E.(Eds) Tratado de Cardiología. Madrid: Elsevier España S.A., 2005: 479-503. [ Links ]
3. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult:A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): Developed in Collaboration With the American College of Chest Physicians and the International Society for Heart and Lung Transplantation:Endorsed by the Heart Rhythm Society.Circulation 2005;112:154-235. [ Links ]
4. Opie LH. The neuroendocrinology of congestive heart failure. Cardiovasc J South Afr 2002;13:171-178. [ Links ]
5. Wright DJ, Tan LB. The role of exercise testing in the evaluation and management of heart failure.Postgrad Med J 1999;75:453-458. [ Links ]
6. Jessup M, Brozena S. Heart Failure. N Engl J Med 2003; 348: 2007-2018. [ Links ]
7. Dávila DF, Núñez TJ, Odreman R, Mazzei de Dávila CA. Mechanisms of neurohormonal activation in chronic congestive heart failure: pathophysiology and therapeutic implications.Int J Cardiol 2005; 101:343-346. [ Links ]
8.Brum PC, Rolim N, Bacurau A, Medeiros A.Neurohumoral activation in heart failure:the role of adrenergic receptors. An Acad Bras Cienc 2006; 78: 485-503. [ Links ]
9. Schrier RW, Abraham WT. Hormones and Hemodinamics in Heart Failure.N Engl J Med 1999; 341: 577-585. [ Links ]
10. Cleland JG, Khand A, Clark A. The heart failure epidemic: exactly how big is it? Eur Heart J 2001; 22: 623-626. [ Links ]
11. Farr MA, Basson CT. Sparking the Failing Heart. N Engl J Med 2004; 351:185-187. [ Links ]
12. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham study. J Am Coll Cardiol 1993; 22: 6A-13A. [ Links ]
13.Goldberg LR,Jessup M.Stage B Heart Failure. Circulation 2006; 113: 2851-2860. [ Links ]
14. Harrington D, Anker SD, Coats AJ. Preservation of exercise capacity and lack of peripheral changes in asymptomatic patients with severely impaired left ventricular function. Eur Heart J 2001; 22:392-399. [ Links ]
15. Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest 2007;117:568-575. [ Links ]
16. Weir R, Mcmurray J, Taylor J, Brady A. Heart Failure in Older Patients. Br J Cardiol 2006; 13: 257-266. [ Links ]
17. Birkeland JA, Sejersted OM, Taraldsen T, Sjaastad I. EC-coupling in normal and failing hearts.Scand Cardiovasc J 2005;13-23. [ Links ]
18. Leite-Moreira AF.Current perspectives in diastolic dysfunction and diastolic heart failure.Heart 2006;92:712-718. [ Links ]
19. Zile, MR. Brutsaert D. New concepts in diastolic dysfunction and diastolic heart failure:part I:diagnosis, prognosis and measurements of diastolic function. Circulation 2002; 105: 1387-1393. [ Links ]
20. Clark AL. Origin of symptoms in chronic heart failure. Heart 2006; 92: 12-16. [ Links ]
21. Ulate G, Ulate A. El calcio en los miocitos cardiacos y su papel en las miocardiopatías. Rev Costarr Cardiol 2006;8:19-25. [ Links ]
22. Benjamin IJ, Schneider MD. Learning from failure:congestive heart failure in the postgenomic age. J Clin Invest 2005; 115: 495-499. [ Links ]
23. Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, et al. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling alter infarction in rats.J Clin Invest 2004;113:727-736. [ Links ]
24. Sande JB, Sjaastad I, Hoen IB, Bokenes J, Tonnessen T, Holt E, et al. Reduced level of serine phosphorylated phospholambn in the failing rat myocardium:A major contributor to reduced SERCA2 activity. Cardiovasc Res 2002; 53: 382-91. [ Links ]
25. Tappia PS. Phospholipid-mediated signaling systems as novel targets for treatment of heart disease. Can J Physiol Pharmacol 2007; 85: 25-41. [ Links ]
26. Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. N Eng J Med 1999; 341: 1276-1283. [ Links ]
27. Swynghedauw B. Phenotypic plasticity of adult myocardium: molecular mechanisms. J Exp Biol 2006; 209: 2320-2327. [ Links ]
28. Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load-induced ventricular hypertrophy progress to systolic heart failure?Am J Physiol Heart Circ Physiol 2005; 289: H8-H16. [ Links ]
29. Teunissen BE, Jongsma HJ, Bierhuizen MF. Regulation of myocardial connexins during hypertrophic remodeling. Eur Heart J 2004; 25: 1979-1989. [ Links ]
30. Watanabe T, Barker TA, Berk BC. Angiotensin II and the endothelium: diverse signals and effects. Hypertension 2005; 45: 163-169. [ Links ]
31. Dorn GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 2005; 115: 527-537. [ Links ]
32. Francis GS, Benedict C, Johnstone DE, Kirlin PC, Nicklas J, Liang CS. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Ventricular Dysfunction (SOLVD). Circulation 1990; 82: 1724-1729. [ Links ]
33. Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and the relation to mortality.CONSENSUS Trial Study Group. Circulation 1990; 82: 1730-1736. [ Links ]
34. Francis GS, McDonald KM, Cohn JN. Neurohumoral activation in preclinical heart failure:remodeling and the potential for intervention. Circulation 1993; 87: IV-90-96. [ Links ]
35. Post SR, Hammond HK, Insel PA. Beta-adrenergic receptors and receptor signaling in heart failure. Annu Rev Pharmacol Toxicol 1999; 39: 343-360. [ Links ]
36. Port JD, Bristow MR. Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol 2001; 33: 887-905. [ Links ]
37. Brum PC, Kosek J, Patterson A, Bernstein D, Kobilka B. Abnormal cardiac function associated with sympathetic nervous system hyperactivity in mice. Am J Physiol Heart Circ Physiol 2002; 283:H1838-H1845. [ Links ]
38. Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res 2003; 93: 896-906. [ Links ]
39. Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of beta(1)-and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation 1999; 100: 2210-2212. [ Links ]
40. Xiao RP, Zhu W, Zheng M, Chakir K, Bond R, Lakatta EG, et al. Subtype-specific beta-adrenoceptor signaling pathways in the heart and the potential clinical implications. Trends Pharmacol Sci 2004; 25: 358-365. [ Links ]
41. Zhao XL,Gutierrez LM, Chang CF, Hosey MM. The alpha 1-aubunit of skeletal muscle L-type Ca channels is the key target for regulation by A-kinase and protein phosphatase-1C.Biochem Biophys Res Commun 1994;198:166-173. [ Links ]
42.Gerhardstein BL,Puri TS,Chien AJ,Hosey MM.Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the beta 2 subunit of L-type voltage-dependent calcium channels. Biochemistry 1999;38:10361-10370. [ Links ]
43.Simmerman HK,Jones LR.Phospholamban:protein structure, mechanism of action,and role in cardiac function.Physiol Rev 1998; 78:921-947. [ Links ]
44.Sulakhe PV,Vo XT.Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli:roles of cyclic nucleotides,calcium,protein kinases and phosphatases and depolarization.Mol Cell Biochem 1995;149-150:103-126. [ Links ]
45.Kunst G,Kress KR,Gruen M,Uttenweiler D,Gautel M,Fink RH. Myosin binding protein C,a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2.Circ Res 2000;86:51-58. [ Links ]
46.Xiao RP.Cell logic for dual coupling of a single class of receptors to G(s)and G(i)proteins.Circ Res 2000;87:635-637. [ Links ]
47.Sibley DR,Strasser RH,Benovic JL,Daniel K,Lefkowitz RJ. Phosphorylation /dephosphorylation of the beta –adrenergic receptor regulates its functional coupling to adenylate cyclase and subcellular distribution.Proc Natl Acad Sci USA 1986;83:94-08-9412. [ Links ]
48.Hausdorff WP,Lohse MJ,Bouvier M,Liggett SB,Caron MG, Lefkowitz RJ.Two kinases mediate agonist-dependent phosphorylation and desensitization of the beta 2-adrenergic receptor.Symp Soc Exp Biol 1990;44:225-240. [ Links ]
49.Ungerer M,Bohm M,Elce JS,Erdmann E,Lohse MJ.Altered expression of beta-adrenergic receptor kinase and beta1-adrenergic receptors in the failing human heart.Circulation 1993;87:454-463. [ Links ]
50.Marx SO,Reiken S,Hisamatsu Y,Jayaraman T,Burkhoff D,Rosemblit N,et al.PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor):defective regulation in failing hearts.Cell 2000;101:365-376. [ Links ]
51.Reiken S,Gaburjakova M,Guatimosim S,Gomez AM,D 'Armiento J, Burkhoff D,et al.Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor)in normal and failing hearts.Role of phosphatases and response to isoproterenol.J Biol Chem 2003;278:444-453. [ Links ]
52.Wehrens XH,Lehnart SE,Marks AR.Intracellular calcium release and cardiac disease.Annu Rev Physiol 2005;67:69-98. [ Links ]
53.Wehrens XH,Marks AR.Altered function and regulation of cardiac ryanodine receptors in cardiac disease.Trends Biochem Sci 2003;28: 671-678. [ Links ]
54.Schwinger RH,Munch G,Bolck B,Karczewski P,Krause EG, Erdmann E.Reduced Ca(2+)-sensitivity of SERCA2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation.J Mol Cell Cardiol 1999;31:479-491. [ Links ]
55.Hajjar RJ,Muller FU,Schmitz W,Schnabel P,Bohn M.Molecular aspects of adrenergic signal transduction in cardiac failure.J Mol Med 1998;76:747-755. [ Links ]
56.Roma G.Catecholamine cardiotoxicity.J Mol Cell Cardiol 1985;17: 291-306. [ Links ]
57.Mann DL,Kent RL,Parsons B,Cooper GT.Adrenergic affects on the biology of the adult mammalian cardiocyte.Circulation 1992;85: 790-804. [ Links ]
58.Levine TB,Francis GS,Goldsmith SR,Simon AB,Cohn JN.Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure.Am J Cardiol 1982;49: 1659-1666. [ Links ]
59.Hensen J,Abraham WT,Dürr J,Schrier RW.Aldosterone in congestive heart failure:analysis of determinants and role in sodium retention. Am J Nephrol 1991;11:441-446. [ Links ]
60.Chun TY,Bloem LJ,Pratt JH.Aldosterone inhibits inducible nitric oxide synthase in neonatal rat Cardiomyocytes. Endocrinology 2003; 144:1712-1717. [ Links ]
61.Fuller PJ,Young MJ.Mechanisms of mineralocorticoid action. Hypertension 2005;46:1227-1235. [ Links ]
62.Ouvrard-Pascaud A,Sainte-Marie Y,Benitah JP,Perrier R,Soukaseum C,Cat AN,et al.Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias.Circulation 2005;111: 3025-3033. [ Links ]
63.Lalevee N,Rebsamen MC,Barrere-Lemaire S,Perrier E,Nargeot J, Benitah JP,et al.Aldosterone increases T-type calcium channel expression and in vitro beating frequency in neonatal rat cardiomyocytes.Cardiovasc Res.2005;67:216-224. [ Links ]
64.Schrier RW.Pathogenesis of sodium and water retention in high-output and low-output cardiac failure,nephrotic syndrome,cirrhosis, and pregnancy.N Engl J Med 1988;319:1065-1072. [ Links ]
65.Raine AEG,Eme P,Burgisser E,Muller FB,Bolli P,Burkart F,et al. Atrial natriuretic peptide and atrial pressure in patients with congestive heart failure.N Engl J Med 1986;315:533-537. [ Links ]
66.Saito Y,Nakao K,Arai H,Sugawara A,Morii N,Yamada T,et al. Atrial natriuretic polypeptide (ANP)in human ventricle:increased gene expression of ANP in dilated cardiomyopathy.Biochem Biophys Res Commun 1987;148:211-217. [ Links ]
67.Ferrari R,Agnoletti G.Atrial natriuretic peptide:its mechanism of release from the atrium.Int J Cardiol 1989;24:137-149. [ Links ]
68.Tsutamoto T,Wada A,Maeda K,Hisanaga T,Maeda Y,Fukai D. Attenuation of compensation of endogenous cardiac natriuretic peptide system in chronic heart failure:prognostic role of plasma brain natriuretic peptide concentration in patients with chronic symptomatic left ventricular dysfunction.Circulation 1997;96:509-516. [ Links ]
69.Olivetti G,Abbi R,Quani F,Kajstura J,Cheng W,Nitahara JA,et al. Apoptosis in the failing heart.N Engl J Med 1997;336:1131-1141. [ Links ]
70.Saraste A,Pulkki K,Kallajoki M,Heikkila P,Laine P,Mattila S,et al. Cardiomyocyte apoptosis and progression of heart failure to transplantation.Eur J Clin Invest 1999;29:380-386. [ Links ]
71.Guerra S,Leri A,Wang X,Finato N,DiLoreto C,Beltrami CA,et al. Myocyte death in the failing human heart is gender dependent.Circ Res 1999;85:856-866. [ Links ]
72.Crow MT,Mani K,Nam YJ,Kitsis RN.The mitochondrial death pathway and cardiac myocyte apoptosis.Circ Res 2004;95:957-970. [ Links ]
73.Wencker D,Chandra M,Nguyen K,Miao W,Garantziotis S,Factor SM,et al.A mechanistic role for cardiac myocyte apoptosis in heart failure.J Clin Invest 2003;111:1497-1504. [ Links ]
74.Zaugg M,Xu W,Lucchinetti E,Shafiq SA,Jamali NZ,Siddiqui MA. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes.Circulation 2000;102:344-350. [ Links ]
75.Zhu WZ,Zheng M,Koch WJ,Lefkowitz RJ,Kobilka BK,Xiao RP. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes.Proc Natl Acad Sci USA 2001;98:1607-1612. [ Links ]
76.Chesley A,Lundberg MS,Asai T,Xiao RP,Ohtani S,Lakatta EG,et al.The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3-kinase.Circ Res 2000;87:1172-1179. [ Links ]
77.Hayakawa Y,Chandra M,Miao W,Shirani J,Brown JH,Dorn GW,et al.Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice.Circulation 2003;108:3036-3041. [ Links ]
78.Adams JW,Sakata Y,Davis MG,Sah VP,Wang Y,Liggett SB,et al. Enhanced Galpha(q)signaling:a common pathway mediates cardiac hypertrophy and apoptotic heart failure.Proc Natl Acad Sci USA 1998;95:10140-10145. [ Links ]
79.Tomaselli GF,Marban E.Electrophysiological remodeling in hypertrophy and heart failure.Cardiovasc Res 1999;42:270-283. [ Links ]
80.Wehrens XH,Lehnart SE,Reiken SR,Deng SX,Vest JA,Cervantes D,et al.Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2.Science 2004;304:292-296. [ Links ]
81.Tomaselli GF,Zipes DP.What causes sudden death in heart failure? Circ Res 2004;95:754-763. [ Links ]
82.Bristow M.Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000;101:558-569. [ Links ]
83.Groenning BA,Nilsson JC,Sondergaard L,Fritz-Hansen T,Larsson HB,Hildebrandt PR.Antiremodeling effects on the left ventricle during beta-blockade with metoprolol in the treatment of chronic heart failure.J Am Coll Cardiol 2000;36:2072-2080. [ Links ]
84.Weber KT.Aldosterone in congestive heart failure.N Engl J Med 2001;345:1689-1697. [ Links ]
85.Gheorghiade M,Teerlink JR,Mebazaa A.Pharmacology of new agents for acute heart failure syndromes.Am J Cardiol 2005;96 [suppl ]: 68G-73G.
* Departamento de Fisiología, Escuela de Medicina, Universidad de Costa Rica
Abreviaturas: ANGII, angiotensina II; ET-11: endotelina 1; GC,gasto cardiaco; HAD, hormona antidiurética (vasopresina);IC,insuficiencia cardiaca; LEC,líquido extracelular; NE,norepinefrina; PA, potencial de acción;PAN, péptido atrial natriurético;PCN, péptido cerebral natriurético; PKA, proteinquinasa A; PKC,proteinquinasa C; RS,retículo sarcoplásmico; RyR2,receptor de rianodina, SERCA2a, ATPasa de calcio de retículo sarcoplásmico;SRAA, sistema renina-angiotensina-aldosterona; VI,ventrículo izquierdo.
Correspondencia: Apartado 1300 - 2050, San Pedro.

