










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.46 suppl.1 San José Oct. 2004
de acromegalia, prolactinomas y
enfermedad de Cushing
Chih Hao Chen-Ku
Asociación Costarricense de Endocrinología, Diabetes y Nutrición
Resumen
Los tumores hipofisiarios funcionantes más frecuentes son los productores de prolactina (prolactinomas), hormona de crecimiento (acromegalia) y ACTH (síndrome de Cushing). Existe mucha controversia con respecto al diagnóstico y manejo de estas patologías. Este artículo presenta una guía para el diagnóstico y tratamiento de tales tumores hipofisiarios, y ha sido elaborado por la Asociación Costarricense de Endocrinología, Diabetes y Nutrición.
La causa más frecuente de acromegalia son los tumores hipofisiarios productores de hormona de crecimiento. El diagnóstico usualmente se retarda varios años luego de haberse iniciado los cambios, por lo lento que se producen las manifestaciones clínicas. El tratamiento inicial de elección consiste en la cirugía, reservándose tratamiento médico con análogos de somatostatina como primera elección, para casos donde ha fallado el tratamiento quirúrgico. Otra posibilidad de tratamiento consiste en la radioterapia, cuya eficacia se ve limitada por el tiempo necesario para hacer efecto.
Con respecto a las hiperprolactinemias, se deben descartar otras causas que produzcan elevación de prolactina. Si se identifica un prolactinoma, el tratamiento va a ser médico en la mayoría de los casos, reservándose la cirugía para aquellos macroprolactinomas con compromiso visual que no han reducido de tamaño con el tratamiento médico. El tratamiento de elección consistió en agonistas dopaminérgicos, y en Costa Rica la elección será bromocriptina, aunque hay otras opciones como cabergolina y quinagolide.
En la enfermedad de Cushing existen diferentes pruebas de diagnóstico, tanto para tamizaje como para diagnóstico confirmatorio. El tratamiento inicial debe ser cirugía, seguida por radioterapia. En este caso el tratamiento médico es mucho menos eficaz que en las otras dos entidades.
Descriptores: acromegalia, prolactinoma, enfermedad de Cushing, guías de tratamiento
Recibido: 30 de enero de 2004 Aceptado: 08 de junio de 2004
Los prolactinomas, acromegalia y enfermedad de Cushing son los tumores funcionantes hipofisiarios que se presentan con mayor frecuencia en la población general. A nivel mundial existe controversia respecto de cómo abordar estas enfermedades, dada la aparición de nuevos tratamientos médicos y otras modalidades de radioterapia. Costa Rica no ha sido la excepción, sobre todo debido a las dudas acerca del manejo de estas patologías hipofisiarias.
Por este motivo, la Asociación Costarricense de Endocrinología Diabetes y Nutrición ha revisado y avalado estas guías para el diagnóstico y manejo de acromegalia, prolactinomas y enfermedad de Cushing, como una forma de unificar los criterios y tratamientos de estas patologías en el ámbito nacional.
Materiales y métodos
Para el presente documento se realizó revisión bibliográfica en Medline, utilizando como palabras claves acromegalia, prolactinomas, síndrome de Cushing y tumores hipofisiarios. De estos artículos se seleccionaron aquellos que fueran de revisión acerca del diagnóstico y tratamiento de los últimos 10 años, y que además estuvieran disponibles en la Biblioteca Nacional de Salud y Seguridad Social (BINASSS), o se encontraran accesibles como referencia de texto completo en Internet, en las páginas de las revistas correspondientes.
Posteriormente, se elaboraró una propuesta inicial de las guías, adaptándolas a los recursos tecnológicos, médicos y terapéuticos disponibles en nuestro medio. Esta propuesta fue discutida en una reunión de la Asociación Costarricense de Endocrinología, Diabetes y Nutrición, en octubre de 2002. Allí se presentaron las correcciones correspondientes, razón por la cual las guías fueron de nuevo discutidas en febrero de este año. Así se logró la aprobación unánime de todos los miembros de la Asociación.
Acromegalia
La acromegalia es una patología ocasionada por la secreción excesiva de hormona de crecimiento. Como consecuencia de esta secreción, se elevan los niveles del factor de crecimiento tipo insulina-1 (IGF-1). La causa más frecuente es el crecimiento de un tumor hipofisiario productor de hormona de crecimiento (98%). En muy raras ocasiones es provocada por la producción ectópica de hormona de crecimiento o factor liberador de hormona de crecimiento por parte tumores pulmonares o pancreáticos, y en ciertas instancias es causada por hiperplasia de las células productoras de hormona de crecimiento 1.
La incidencia de la acromegalia en el ámbito mundial es de 3-4 casos por millón de habitantes por año, con una prevalencia de 50-70 casos por millón de habitantes 2. En Costa Rica no hay datos nacionales sobre su epidemiología, aunque contamos con alrededor de 80 pacientes tratados en los servicios de endocrinología de los hospitales San Juan de Dios, México y Calderón Guardia (datos personales del autor no publicados).
a. Manifestaciones clínicas
La acromegalia por lo general se diagnostica entre 10 y 15 años después del inicio de los síntomas, debido a que estos se presentan de una forma lenta y progresiva. Se estima que en un 40% de casos, el diagnóstico se hace al consultar el paciente por otros motivos. Los síntomas pueden dividirse entre los que se deben a la expansión del tumor y aquellos relacionados con la secreción excesiva de hormona de crecimiento 1.
Los síntomas relacionados con la expansión del tumor son cefalea, pérdida de campos visuales periféricos (hemianopsia bitemporal), destrucción del tejido hipofisiario adyacente, que produce los diferentes síntomas del panhipopi-tuitarismo 3.
Los síntomas relacionados con el exceso de hormona de crecimiento son rasgos faciales más toscos, prominencia de arcos ciliares, prognatismo, macroglosia, crecimiento acral de manos y pies, así como edema de dedos, manos y pies, sudoración excesiva, osteoartrosis, síndrome de túnel carpal y apnea obstructiva de sueño 3.
La acromegalia produce una serie de trastornos metabólicos, que incluyen intolerancia a la glucosa o diabetes mellitus, hipertensión arterial, cardiomegalia con hipertrofia ventricular izquierda, y enfermedad arterial coronaria como consecuencia de lo anterior 1.
Al diagnosticarse un paciente con acromegalia, éste tiene una mortalidad de 2 a 3 veces mayor a la de la población general. Las causas de mortalidad, en orden de frecuencia, son enfermedad arterial coronaria, problemas respiratorios, y cáncer 4.
Existe gran controversia entre la relación de acromegalia y cáncer. Lo que sí se conoce con certeza es el vínculo con pólipos colónicos 5, aunque no necesariamente lleguen a malignizar. En algunas series se ha reportado que el riesgo relativo de cáncer de colon es hasta 29 veces mayor con respecto a la población general. Las series son todavía más controversiales al relacionarlos con el del cáncer gástrico o esofágico.
b. Diagnóstico
Para realizar el diagnóstico, se requiere en primera instancia, la sospecha clínica del paciente, quién suele ser referido a un servicio de endocrinología, por un médico que ha visto el fenotipo del paciente cuando consulta por algún otro síntoma, como por ejemplo, diabetes mellitus, túnel carpal o incluso osteoartrosis. Esta sospecha clínica debe confirmarse con pruebas que demuestren la hipersecreción de hormona de crecimiento.
La prueba de tamizaje inicial puede efectuarse con niveles de factor de crecimiento tipo insulina-I (IGF-I) ó somatomedina C y con niveles de hormona de crecimiento basal. Los niveles de IGF-1 deben compararse con los valores normales para la edad del paciente. En los pacientes acromegálicos, el tener hipersecreción de hormona de crecimiento, estimula al hígado a que produzca niveles altos de IGF-I, que al final son los que median los efectos clínicos observados 1.
Hay diversas pruebas dinámicas para demostrar la secreción autónoma de hormona de crecimiento, sin embargo, la más aceptada en estos momentos es la curva de tolerancia a la glucosa.
La curva de tolerancia a la glucosa se realiza en estado de ayuno. Se coloca un catéter intravenoso en el paciente, luego se administra una carga de 75 g de glucosa vía oral, posterior a lo cual se toman niveles de glucosa y hormona de crecimiento basal, cada 30 minutos, hasta las 2 horas. La falta de supresibilidad de los niveles de hormona de crecimiento a <1 ng/ml, o la presencia de una elevación paradójica de esta, confirma el diagnóstico 6. Existe controversia mundial sobre si esta prueba debe realizarse en pacientes diabéticos, dada la hiperglicemia que puede llegar a producirse al realizar la curva de tolerancia.
Por otro lado, niveles basales de hormona de crecimiento < 2 µg/dl predicen que la respuesta en la curva de tolerancia llegará niveles de <1 µ/dl, por lo que puede obviarse la curva de tolerancia.
Otras pruebas dinámicas que pueden ayudar en caso de duda, incluyen la respuesta de la hormona de crecimiento a la aplicación de TRH o a la infusión de dopamina, pero por lo general no son necesarios.
c. Estudios de localización
Una vez confirmada la secreción autónoma de hormona de crecimiento, se debe localizar la fuente de esta hipersecreción. Como se dijo anteriormente, el 98% de los casos son causa de un adenoma productor de hormona de crecimiento. Pero, hay un 2% de los casos que pueden ser causados por otros tumores y este es el porcentaje que se debe descartar con los estudios de localización 1.
Si se dispone, el estudio de elección, es la resonancia magnética nuclear, ya que tiene mayor sensibilidad. En Costa Rica este recurso no es tan accesible y no se cuenta con ella en el Seguro Social, por lo que el estudio de imagen de elección corresponde a la tomografía axial computarizada (TAC). Si el TAC demuestra la presencia de un adenoma hipofisiario, se puede afirmar que ese adenoma es el productor de hormona de crecimiento, aunque idealmente esto se debe acompañar de niveles suprimidos de GHRH (hormona liberadora de hormona de crecimiento). Por otro lado, si el TAC no logra demostrar la presencia de adenoma hipofisiario, se debe proceder a la realización de resonancia magnética nuclear, por las características descritas.
En caso de que la resonancia magnética nuclear no logre demostrar la presencia de un adenoma hipofisiario, se debe proceder a descartar la producción ectópica de la hormona de crecimiento o de la hormona liberadora de hormona de crecimiento. En estos casos las causas más frecuentes son tumores neuroendocrinos en pulmón o páncreas, por lo que se debe practicar una tomografía axial computarizada de tórax y abdomen, en busca de estos tumores.
Una alternativa a los estudios por imágenes es la medición de la hormona liberadora de hormona de crecimiento, pero tiene la limitante de que no se realiza en el país de forma rutinaria (aunque se puede contactar con laboratorios privados para el envío de muestras al exterior), y por lo tanto se hace difícil su implementación. En caso de efectuarse la medición, si los niveles están bajos o suprimidos, orienta a producción de hormona de crecimiento, mientras que si están elevados orienta a tumor productor de GHRH, y debido a esto se debe buscar respuesta con tomografía de tórax y abdomen, en el sitio de la producción ectópica de esta.
d. Tratamiento
Es de consenso que el tratamiento inicial debe consistir en la cirugía, ya que es el método que ofrece mejores tasas de curación. El abordaje quirúrgico depende de la situación de cada paciente y de lo extenso de la lesión tumoral. Las mejores series a nivel mundial reportan tasas de curación del 70% para microadenomas (tumores que miden menos de 10 mm en su diámetro mayor), y hasta de un 40% para macroadenomas (tumores que miden más de 10 mm en su diámetro mayor). Sin embargo, las tasas de curación varían según los criterios utilizados. Desafortunadamente no hay datos nacionales para compararlos 7-8.
En caso de que el paciente presente alguna contraindicación para la cirugía, como cardiopatía descompensada, en ciertos lugares se ha planteado el manejo médico inicial con análogos de somatostatina (octréotido ó lanreótido) para tratar de mejorar el estado general del paciente y que sea luego sometido a la cirugía operación 9-11.
Seis semanas después de la cirugía se deben valorar los criterios de curación. Nuevamente, se recomienda realizar una curva de tolerancia a la glucosa con 75 g de esta, para medir los niveles de hormona de crecimiento, con las consideraciones del caso en pacientes diabéticos.
El acuerdo en este momento es utilizar los criterios del consenso de Cortina, que indican que los niveles deben suprimirse a <1 µg/dl, y tener los niveles de IGF-I normales para la edad 5.
De no alcanzarse criterios de curación, se debe recurrir a terapias alternativas. Una de estas es la radioterapia 12, con cobaltoterapia convencional o con gamma knife. En ambas instancias la respuesta que se observa no es inmediata, gamma knife tiene resultados más pronto, respecto de la cobaltoterapia. Debido a lo anterior, a pesar de haber recibido radioterapia, el paciente debe ser candidato a terapia médica, mientras la radioterapia hace efecto.
La terapia médica de elección, por tolerancia y efectividad, son los análogos de somatostatina (octreótido 13 y lanreotido de depósito 11,13). En el medio se cuenta únicamente con octreótido. La aplicación de octréotido depósito logra llevar a tasas de curación de hasta 50-70%. Los efectos de esta terapia persisten por un período de hasta 28 días. Antes de aplicar la siguiente dosis es conveniente valorar los niveles de hormona de crecimiento IGF-I, para ver si se cumplen los criterios de curación. En caso de no hacerlo, se puede aumentar la dosis o frecuencia de administración. Si se alcanzan los criterios de curación, se debe continuar con el tratamiento con análogos de somatostatina hasta esperar que la radioterapia haga efecto y es preciso valorar periódicamente los niveles de hormona de crecimiento e IGF-I.
De no lograr alcanzar los criterios de curación con los análogos de somatostatina, se deben plantear otras terapias. Una de las posibilidades es una nueva cirugía, aunque se sabe que las reintervenciones conllevan una mayor morbilidad. Otra opción disponible son los análogos de dopamina 14 (bromocriptina, cabergolina, pergolide), sin embargo, tienen poca tolerancia y su eficacia, por lo general, es <40% 15. Esta terapia médica es más efectiva en caso de ser adenomas que cosecretan prolactina, aparte de hormona de crecimiento.
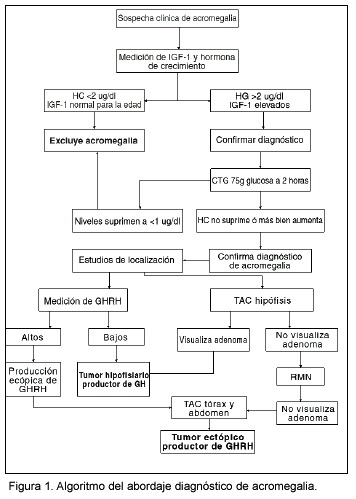
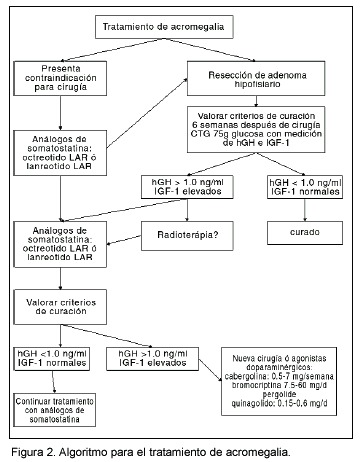
Las figuras 1 y 2 presentan algoritmos para el diagnóstico y tratamiento de la acromegalia.
Prolactinoma
a. Manifestaciones clínicas
Los síntomas producidos por los prolactinomas también pueden dividirse en los generados por compresión del tumor y los relacionados con el exceso de prolactina. El aumento de prolactina conlleva a un estado de hipogonadismo, tanto en hombres como mujeres. Por lo tanto, en el hombre, una de las manifestaciones será la disfunción eréctil, y en las mujeres, se manifestará de forma más temprana, como oligomenorreas o amenorreas asociadas a galactorrea 16.
b. Abordaje diagnóstico
En el paciente con diagnóstico clínico y bioquímico de hiperprolactinemia, se deben descartar, en primera instancia, causas fisiológicas y secundarias de elevación en los niveles de prolactina.
Entre las causas fisiológicas, se pueden citar el embarazo y la lactancia. Entre las farmacológicas, se deben descartar: el uso de psicotrópicos, estrógenos, antagonistas dopaminérgicos como metoclopramida, antidepresivos tricíclicos, cocaína, alfametildopa, verapamilo e inhibidores de bomba de protones. También, se deben descartar otras causas secundarias, como el hipotiroidismo, e insuficiencia renal crónica 17, y estímulos repetitivos o manipulación del área mamaria.
Descartadas las causas secundarias, se deben realizar estudios de imágenes de hipófisis para valorar el tamaño del adenoma hipofisiario. En este caso, el estudio de mayor sensibilidad es la resonancia magnética nuclear 17.
En el país, al ser tan limitado el acceso a la resonancia magnética nuclear, se debe recurrir a el TAC. El TAC puede dar una idea bastante clara sobre el prolactinoma, ya que al ser una patología predominantemente de manejo médico, no es tan importante valorar la presencia de microadenomas. Por otra parte, en la mayoría de las ocasiones, cuando se trata de macroadenomas con compromiso visual, se logra detectarlos mediante este recurso.
Los estudios por imágenes pueden revelar que la hipófisis es normal, en cuyo caso se catalogaría como hiperprolactinemia idiopática, ya que hasta ahora no está descrita la producción ectópica de prolactina 16, aunque no se descarta la presencia de microadenomas hipofisiarios no identificados por estudios de imágenes.
Si se logra evidenciar la presencia de un adenoma hipofisiario, se deben valorar los niveles de prolactina. Se ha observado que los niveles de prolactina correlacionan con el tamaño tumoral y la mayoría de los prolactinomas los tienen superiores a 150 ng/ml. Se debe tomar en cuenta que la compresión del tallo hipofisiario, con la interrupción de la transmisión de la dopamina como factor inhibidor de secreción de prolactina, puede generar elevaciones de prolactina, sin que el tumor sea la fuente de producción. Por lo tanto, se debe considerar esta posibilidad en los casos donde los niveles de prolactina son menores a 150 ng/ml; y descartar que la elevación de prolactina sea secundaria a un tumor hipofisiario de otra etiología 17.
En ambos casos, independientemente del nivel de prolactina, se recomienda realizar pruebas para valorar la reserva hipofisiaria. El test de elección sigue siendo la prueba de tolerancia a la insulina. Como es de suponer, si hubiera alguna deficiencia absoluta o parcial de algunos de los ejes, se debe sustituir con las hormonas correspondientes.
c. Tratamiento
En primer lugar, el principal determinante para decidir la vía del tratamiento inicial será la presencia o no de compromiso visual. Si el tumor se ha expandido y ha habido compresión quiasmática que comprometa la viabilidad del nervio óptico, el tratamiento inicial deben ser los agonistas dopaminérgicos, pero con la precaución de un seguimiento estrecho del paciente. El tratamiento inicial es médico, gracias a que éste logra reducir el tamaño tumoral, y por lo tanto, en algunos casos, consigue también la reducción de la compresión quiasmática 18. Después de cuatro meses de evaluación del tamaño tumoral, se debe indicar un TAC. De no lograrse la reducción del tumor hipofisiario, se debe proceder al tratamiento quirúrgico 19 para realizar la descompresión del nervio. Este tratamiento no debe ser el inicial en aquellos pacientes sin compromiso visual, ya que la tasa de curación es baja y la recidiva es alta.
Tras la cirugía se debe revalorar los niveles de prolactina, si se han normalizado se puede considerar que el paciente ha sido curado, mientras que si persisten alto, se indica tratamiento médico.
El tratamiento médico es de elección en aquellos pacientes donde no haya compromiso visual o que la cirugía no ha logrado curar 20, 21, lo constituyen los agonistas dopaminérgicos. El agonista dopaminérgico inicial en el país es la bromocriptina, ya que este agente es efectivo, posee mayor cantidad de estudios que apoyan su efectividad y es de menor costo económico; de esta forma se logra alcanzar tasas de normalización de prolactina similares a las de los otros agentes. El problema con la bromocriptina es su tolerabilidad, por lo que en presencia de reacciones adversas importantes, se aconseja proceder a agonistas dopaminérgicos alternativos, como por ejemplo, la cabergolina, quinagolide y pergolide. Se debe hacer la salvedad de que puede haber falla terapéutica a la bromocriptina, en cuyo caso estos otros agentes, y especialmente la cabergolina, pueden ser efectivos 22. Una forma de mejorar la tolerancia gastrointestinal de la bromocriptina es la administración intravaginal de las tabletas orales, con la misma efectividad.
En caso de haber falla terapéutica o intolerancia a los agonistas dopaminérgicos, como último recurso se puede recurrir a la radioterapia, aunque se sabe su la efectividad es a muy largo plazo y la tasa de curación es baja.
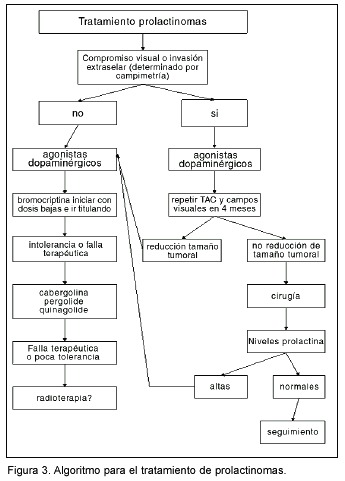
La figura 3 muestra un algoritmo donde se resume el manejo de los prolactinomas.
Síndrome de Cushing
El síndrome de Cushing se produce por un estado crónico de hipercortisolismo, que afecta casi la totalidad de los tejidos del organismo 23.
La incidencia para el síndrome de Cushing espontáneo es de aproximadamente 2-4 casos por millón de habitantes, por año. Sin embargo, debe considerarse que la prevalencia real es mucho mayor, debido a que la etiología más frecuente es el iatrogénico 24.
a. Etiología
La clasificación etiológica divide al síndrome de Cushing en dos grandes grupos, según exista o no un exceso de ACTH a nivel plasmático. En el síndrome de Cushing ACTH dependiente 25, se tiene la enfermedad de Cushing que se produce por aumento de ACTH secretado por la hipófisis, con hiperplasia adrenocortical bilateral secundaria. Por otro lado, puede ser por producción de ACTH ectópico, ya sea producido por secreción de ACTH o péptidos similares a ACTH, por tumores extrahipofisiarios Por último, puede ser iatrogénico por administración de cantidades excesivas de ACTH o de análogos sintéticos.
El síndrome de Cushing ACTH independiente 25 puede ser de origen suprarrenal, originado por adenoma o carcinoma suprarrenal productor de cortisol; con menor frecuencia, por hiperplasia adrenal bilateral. También puede producirse de forma iatrogénica, por administración suprafisiológica prolongada de corticosteroides.
La frecuencia de las diferentes etiologías varía según la serie que se revise. Se considera que la causa más frecuente es la administración exógena de corticosteroides 26. De los casos de síndrome de Cushing endógenos, se calcula que entre un 65-75% corresponden a enfermedad de Cushing, 10-15% corresponden al síndrome de ACTH ectópico y un 10% es de origen suprarrenal.
En aproximadamente un 80-85% de los casos de enfermedad de Cushing, se encuentra un microadenoma. En menos del 10% de casos se halla un macroadenoma. Alrededor del 5% presenta hiperplasia difusa de células corticotropas 26.
La mayoría de los casos de síndrome de Cushing adrenal, son producidos por un adenoma. Sin embargo, la diferenciación entre adenoma y carcinoma por criterios histológicos muchas veces es difícil. Por lo general, los carcinomas adrenales son más voluminosos, con pesos mayores a 100 gramos, mientras que los adenomas tienden a pesar entre 10 y 70 gramos. La mayoría de los autores diagnostican carcinoma adrenal solo ante la presencia de invasión capsular, vascular o metástasis a distancia.
En la producción ectópica de ACTH, los tumores en orden de frecuencia son: carcinoma broncogénico, carcinoide-especialmente bronquial, timoma, adenoma bronquial, tumor pancreático y carcinoma medular de tiroides.
b. Manifestaciones clínicas:
Las principales manifestaciones clínicas que se presentan son 26-27: obesidad centrípeta (88%), hipertensión arterial (78%), plétora (78%), alteraciones en la reproducción y función sexual (68%), hirsutismo y virilización (66%), debilidad muscular (61%), sangrado fácil (hematomas) (51%), osteoporosis (51%), acné (49%), estrías rojas o purpúreas (46%), alteraciones afectivas (42%) y edemas (38%).
De todas estas, el síntoma de mayor sensibilidad será un aumento súbito de peso (como obesidad central). Otras manifestaciones que ayudan en este sentido son cara luna llena, debilidad muscular proximal, cambios en piel o la presencia de osteoporosis sin explicación, hipertensión arterial, resistencia a la insulina (son poco sensibles dada su alta prevalencia en la población general), afección del tercer par craneal (se observa hasta en un 25% de casos de enfermedad de Cushing, a pesar de que muchas veces clínicamente no se busca o no se reconoce). El hallazgo más común en niños es el retraso o detención de crecimiento.
La presencia de un inicio súbito de la sintomatología orienta más a origen ectópico de ACTH, mientras que la instauración paulatina sugiere enfermedad de Cushing.
c. Diagnóstico:
Existen pruebas diferentes para establecer el hipercortisolismo y tratar de determinar el origen de esta producción. Hay que tomar en cuenta que en la enfermedad de Cushing se mantiene el retrocontrol sobre la producción de ACTH por la hipófisis, pero que requiere dosis mayores (suprafisiológicas) de corticosteroides.
Cualquier prueba que se emplee, por ejemplo, la de tamizaje, presentará el problema de que hay condiciones clínicas más frecuentes que conducirán a la producción de hipercortisolismo. Además, para alcanzar una sensibilidad diagnóstica cercana al 100%, muchas de estas pruebas tendrán una baja especificidad y provocarán una gran cantidad de falsos positivos. Por lo tanto, debe aumentarse la posibilidad pretest, al realizar el tamizaje en pacientes con sospecha clínica.
Antes de efectuar cualquier prueba bioquímica, el clínico debe tener en cuenta patologías que llevan a hipercortisolismo, sin producir síndrome de Cushing.
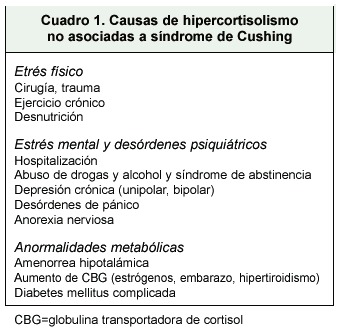
Después de descartar fuentes exógenas de esteroides, la causa más frecuente de hipercortisolismo será la presencia de patología psiquiátrica o estrés físico o mental. Un 25% de los pacientes hospitalizados y un 33% de los pacientes en el postoperatorio inmediato, tendrán niveles altos de cortisol. El paciente alcohólico debe abstenerse al menos uno o dos meses, antes de realizar las diferentes pruebas 28. El cuadro 1 muestra situaciones donde hay aumento en los niveles de cortisol, que no se deben al síndrome de Cushing.
d. Test de tamizaje:
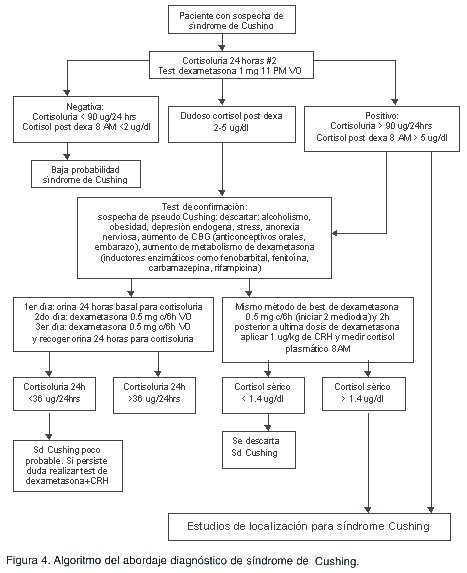
Existen varias pruebas que se pueden realizar, como tamizaje, para excluir la producción de hipercortisolismo. En esta instancia, por la facilidad, sensibilidad y especificidad, se postula que la cortisoluria de 24 horas y el test de supresión con dexametasona de 1 mg, son los de elección 29.
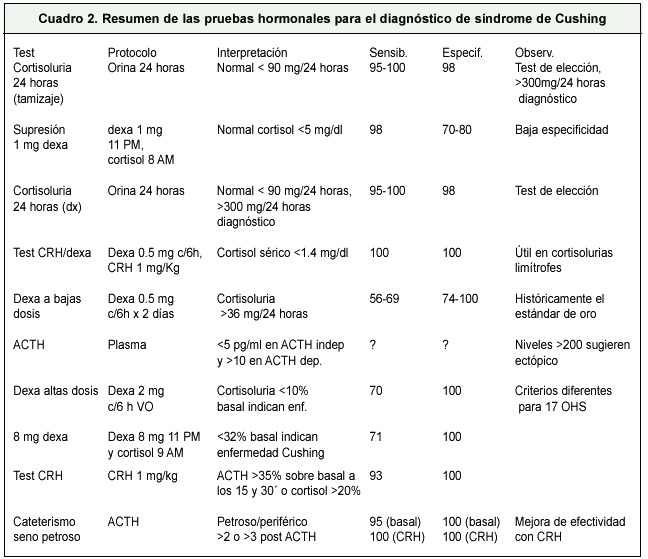
Cortisoluria de 24 horas: Se basa en la medición de la excreción urinaria de cortisol libre en 24 horas. Se recoge a las 7 a.m, cuando el paciente elimina la primera orina de la mañana. A partir de entonces se recoge la orina hasta las 7 a.m. del día siguiente (incluyendo esta última). Esta muestra debe guardarse en una hielera, hasta el momento de la determinación. Es el test más sensible (95-100%) y específico (98%). Se debe comprobar que la recolección de orina ha sido adecuada, midiendo la excreción de creatinina (normal 1-1,5 gramos en 24 horas). La sensibilidad aumenta a más del 98%, si se recoge más de 1 orina de 24 horas. Aunque los valores pueden variar según el tipo de prueba que se use, se considera que valores superiores a 250-300 mg 24 horas, son virtualmente diagnósticos de síndrome de Cushing. Sin embargo, debe prevalecer el criterio clínico, en especial si se sospecha secreción intermitente de cortisol 30.
Test de supresión con 1 mg de dexametasona: En esta prueba se administra 1 mg de dexametasona VO a las 11 p.m, y el nivel de cortisol plasmático se mide al día siguiente, a las 8 a.m. La dexametasona no interfiere con el método de laboratorio usado para medir cortisol, lo que permite valorar la supresibilidad de la producción endógena de cortisol. La prueba se basa en el principio de que prácticamente todas las fuentes de producción inapropiada de cortisol o ACTH, no se inhiben con 1 mg de dexametasona, manteniendo niveles mayores de 5 mg/dl, 9 horas posteriores a la administración. Tiene una sensibilidad del 98%, pero con una especificidad de entre el 70-80%. La sensibilidad de la prueba se ve afectada por cualquier condición donde se altere el aporte de dexametasona, lo que podría producir resultados positivos falsos. Deben considerarse, por lo tanto, factores como inductores hepáticos (fenitoína, carbamazepina, fenobarbital, rifampicina), y niveles altos de CBG (globulina transportadora de cortisol). Por otro lado, condiciones como hepatopatía crónica o insuficiencia renal crónica llevan a falsos negativos, al eliminar en mayor tiempo la dexametasona. Excluyendo estas condiciones, el test de dexametasona es el más útil, ya que es mucho más fácil de realizar, pero al combinarlo con la cortisoluria de 24 horas y con un resultado negativo en ambas pruebas, se puede descartar la presencia de síndrome del Cushing 27.
Cortisol sérico a medianoche: En ciertos estudios se ha demostrado que, obteniendo una muestra a las 12 mn para cortisol, a través de un catéter colocado previamente y sin provocar dolor ni despertar al paciente, se puede excluir el síndrome de Cushing con una sensibilidad del 98%. Los pacientes con síndrome de Cushing tienden a tener niveles mayores de 1.8 mg/dl. Sin embargo, las dificultades en la obtención de la muestra y la necesidad de que el paciente sea internado, hacen de esta prueba poco práctica 31.
Otras pruebas: Las otras pruebas descritas no se deben emplear para tamizaje. Los niveles séricos de cortisol y ACTH al azar no sirven para establecer diagnóstico, dado su ciclo circadiano. La medición de cortisol en saliva tiene la ventaja de que determina el nivel de cortisol libre, sin ser modificado por los niveles de CBG, pero la experiencia con esta prueba es poca y la casuística acumulada hasta ahora no permite hacer recomendaciones para su uso rutinario.
e. Diagnóstico definitivo:
Cuando la cortisoluria 24 horas se encuentra en el límite y hay sospecha de síndrome de Cushing, amerita las pruebas confirmatorias.
Cortisoluria 24 horas como test confirmatorio: En pacientes que recolecten adecuadamente la orina, muchos autores lo consideran el test confirmatorio de elección. La excreción urinaria de 17 cetosteroides se empleó anteriormente como test confirmatorio, pero ha caído en desuso dada su baja sensibilidad y su alta probabilidad de interferencia en la medición, al utilizarse con ciertos medicamentos. En los casos de secreción intermitente de cortisol, se recomienda la medición seriada de cortisolurias de 24 horas, por lo menos una vez a la semana, por entre 3 y 6 meses, para poder descartar el diagnóstico 30.
Test de supresión con CRH/dexametasona: Esta prueba se basa en 2 observaciones: la secreción de cortisol por causas no relacionadas al síndrome de Cushing, se puede suprimir con dexametasona en bajas dosis, y la respuesta hipofisiaria a la CRH suele estar suprimida, dadas los niveles altos de cortisol (en pacientes con hipercortisolismos no Cushing). El test consiste en administrar dexametasona 0.5 mg cada 6 horas, vía oral, con la última dosis a las 6 a.m. (12 md, 6 p.m.; 12 mn, 6 a.m.; 12 md, 6 p.m.; 12 mn y 6 a.m.) Posteriormente, a las 8 a.m, por un bolo de CRH (1mg/kg peso corporal), midiendo el nivel de cortisol plasmático 15 minutos después. Al usar un nivel corte de cortisol en 1.4 mg/dl, este test tiene una sensibilidad y especificidad prácticamente del 100%, en una población seleccionada (alta probabilidad pretest). Este test establece el diagnóstico del síndrome de Cushing, independientemente de la causa, por lo que se usa como test confirmatorio, ya que se pueden diferenciar las hipercortisolemias por otras causas de los síndromes de Cushing verdaderos. Sin embargo, existe la gran limitante de que en Costa Rica no se cuenta con CRH 32.
Test de supresión con dexametasona en dosis bajas: El test de Liddle se consideró el estándar de oro contra el cual se comparaban las otras pruebas. Consiste en la recolección de 2 orinas de 24 horas basales y la administración de dexametasona 0.5 mg VO cada 6 horas, por 48 horas. Durante las segundas 24 horas, de administración de dexametasona, se recolecta una nueva orina de 24 horas para determinar la cortisoluria. Si la excreción urinaria de cortisol, en las últimas 24 horas, es mayor de 36 mg/24 horas, se considera positivo para el síndrome de Cushing. Pero, el test tiene una sensibilidad del 56%, con una especificidad del 100% 32.
f. Pruebas de localización
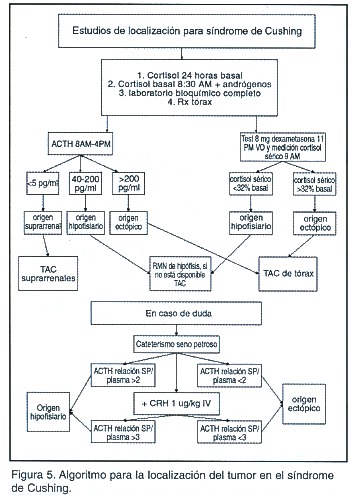
Una vez confirmado el síndrome de Cushing, la pregunta que se debe responder es la fuente de producción del cortisol que hay en exceso.
Niveles de ACTH plasmático: La determinación de un nivel de ACTH basal permite distinguir si el síndrome de Cushing es ACTH dependiente o no. Se considera ACTH dependiente, si el nivel es >10 pg/ml, e independiente si es <5 pg/ml. Para niveles intermedios (5-10 pg/ml), la causa puede ser tanto en ACTH dependiente como ACTH independiente, por lo que está indicado el test con CRH. La sensibilidad y especificidad de una determinación aislada de ACTH no están bien determinadas. Otros estudios reportan que el nivel del ACTH puede orientar, ya que niveles por encima de 200 pg/ml, sugieren, producción ectópica, mientras que niveles de 40-200 pg/ml son compatibles más bien con enfermedad de Cushing 33.
Test de supresión con altas dosis de dexametasona: En pacientes con ACTH alto, se debe distinguir el origen, ya sea hipofisiario o ectópico. Este test se basa en la presunción de que la secreción de ACTH de origen hipofisiario puede suprimirse con altas dosis de dexametasona, mientras que la del ectópico no lo hace. El test se efectúa al recolectar una orina de 24 horas basal (con medición de cortisoluria), iniciando luego dexametasona, 2 mg cada 6 horas, vía oral, por 48 horas, y recogiendo una nueva orina de 24 horas en el segundo día de administración de dexametasona. Con el criterio usado al principio por Liddle (disminución en el 50% de los niveles de 17 hidroxiesteroides), el test tenía una sensibilidad del 80-85% y una especificidad del 70-90%. Sin embargo, si se usa el criterio de supresión de más del 90% de la cortisoluria, da una sensibilidad del 70%, con una especificidad del 100%. Pero un, 10% de tumores, especialmente carcinoides, se pueden suprimir con este test. Se ha visto también que hasta el 92% de los microadenomas se suprimen con este test, mientras que tan solo un 56% de los macroadenomas lo hacen 34.
Test de supresión overnight con 8 mg de dexametasona: En esta prueba se obtiene un nivel basal de cortisol sérico a las 8 a.m., seguido de una administración de 8 mg de dexametasona a las 11 p.m., y una nueva muestra para cortisol a las 8 a.m. del día siguiente. Se usa el criterio de que el nivel de cortisol debe suprimir más del 50%, con lo que hay una sensibilidad del 88-92% y una especificidad del 100%. Al emplearse esta prueba se deben tener en cuenta siempre los errores de interpretación, que le pueden conferir un metabolismo alterado de la dexametasona 35.
Test de CRH: Esta prueba se basa en que una fuente de producción hipofisiaria responde ante CRH; sucede diferente si es de origen ectópico o suprarrenal. Por lo tanto, es de utilidad en el paciente en donde los niveles de ACTH son limítrofes, o se precisa diferenciar entre enfermedad de Cushing y producción ectópica de ACTH. En este test se mide un nivel de ACTH basal, luego se administra CRH 1 mg/kg y se miden niveles de ACTH a los 15 y 30 minutos. Si el aumento del ACTH es de más del 35%, sugiere enfermedad de Cushing, con una sensibilidad del 93% y una especificidad del 100%. También se puede determinar el nivel de cortisol basal, 30 y 45 minutos post CRH, donde si aumenta más del 20%, sugiere enfermedad de Cushing, con sensibilidad del 91% y especificidad del 88%. En el 10-20% de los pacientes que se realizan esta prueba, se presentará flushing facial 36.
Cateterismo de seno petroso inferior: Muchos centros consideran que es esencial en el estudio del síndrome de Cushing, sobre todo en pacientes sin lesión tumoral en el sitio que indican los test bioquímicos. Se toma una muestra de sangre de ambos senos petrosos y de sangre periférica. En el paciente con enfermedad de Cushing, se obtiene un gradiente de ACTH seno petroso/periférico >2, y >3, si se estimula con CRH (1 mg/kg peso). Si el gradiente entre un lado y otro es >1.4, se puede lateralizar la localización hasta en un 80%37.
Estudios por imágenes: Se relega al último lugar, por cuanto el 10-20% de la población tiene incidentalomas suprarrenales o hipofisiarios. Si la sospecha es una lesión hipofisiaria, el test de elección es la resonancia magnética, al tener una mayor sensibilidad, ya que el TAC identificará tan solo 50% de los casos. De haber secreción ectópica, el primer sitio que se debe buscar es con TAC de tórax, seguido de abdomen. La gamagrafía con octreótido puede ayudar a localizar los tumores productores de ACTH.
El Cuadro 2 muestra un resumen de los estudios hormonales que se pueden realizar para el diagnóstico del síndrome de Cushing y la Figura 4 señala un algoritmo para el abordaje diagnóstico de esta patología. La Figura 5 esquematiza la forma de localizar el tumor productor del síndrome de Cushing.
g. Tratamiento
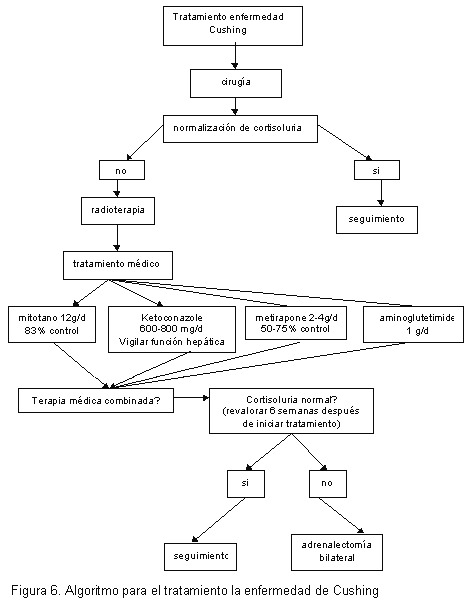
El tratamiento de elección que ofrece mayores posibilidades de curación es la cirugía. La tasa de curación depende del tamaño del adenoma hipofisiario y de la experiencia del centro hospitalario, en caso de que exista enfermedad de Cushing. Cuando se trata de adenomas suprarrenales, la cirugía también constituye la primera elección con tasas de curación mayores. En los casos más raros de secreción ectópica de ACTH, la vía quirúrgica es de elección, aunque el pronóstico es reservado, por la agresividad del tumor primario 38.
Luego de la cirugía se debe revalorar la cortisoluria a las 6 semanas. En caso de normalizarse, se considera que el paciente ha entrado en curación. De lo contrario, se deben recomendar otras vías terapéuticas, como la radioterapia 39. Al igual que en otros tumores hipofisiarios, este procedimiento no tiene efecto de forma inmediata, por lo que se debe recurrir a alternativas de terapia, mientras hace efecto la radioterapia; es en esta instancia donde el tratamiento médico puede ser considerado.
El tratamiento médico consiste en terapia orientada a inhibir la síntesis de glucocorticoides; en este sentido, se cuenta con varios agentes.
La metirapona inhibe la 11 hidroxilasa y produce reducciones rápidas en los niveles endógenos de cortisol, que pueden ocurrir incluso en las primeras dos horas. Las dosis requeridas varían entre 500 y 6000 mg/día, y se deben titular según los niveles de cortisol. Dentro de sus efectos secundarios habituales están el acné y el hirsutismo, y además, en algunas ocasiones se puede producir hipertensión, edema e hipokalemia. Este agente no se encuentra disponible en Costa Rica.
El ketoconazole es un antifúngico imidazole que inhibe la 11 hidroxilasa, la 17-20 liasa, la 17 hidroxilasa y la 18 hidroxilasa. La dosis habitual inicial es de 200 mg bid, y el inicio del efecto es más retardado que el de la metirapona. El efecto adverso principal que limita la utilización del ketoconazole es la hepatotoxicidad, con elevación reversible de transaminasas, hasta en el 5-10% de pacientes (y con lesión hepática severa en 1/15.000 pacientes). Se debe estar siempre pendiente de la posibilidad de inducción de insuficiencia suprarrenal. Dada su capacidad de inhibir la síntesis de andrógenos, se constituye la elección en mujeres con hirsutismo asociado; sin embargo, este mismo efecto puede llevar a hipogonadismo en hombres.
El aminoglutetimide inhibe la conversión de colesterol a pregnenolona, la 18 hidroxilasa, 11 hidroxilasa y la aromatasa. Este agente es menos efectivo para tratarla enfermedad de Cushing, en comparación con las otras causas de hipercortisolismo. Sus efectos secundarios frecuentes constituyen rash, fiebre, mareos y letargia, los cuales que se pueden presentar hasta en un 58% de los pacientes.
El mitotano inhibe la enzima que convierte el colesterol en pregnenolona y, además, la 11 hidroxilasa. Tiene un lento inicio de acción, pero sus efectos persisten una vez suspendido el tratamiento. Hasta un 57% de los pacientes que lo toman, llegan a tener efectos adversos gastrointestinales, alteraciones neurológicas, elevación de transaminasas, hipercolesterolemia, ginecomastia y prolongación de tiempos de coagulación.
En algunas circunstancias se ha intentado el uso de terapia médica combinada, para reducir las dosis de los diferentes agentes utilizados y también los efectos adversos.
Pero, hay muy poca experiencia en este sentido y se recomienda individualizar cada caso. Por otro lado, en Costa Rica solo se encuentra con ketoconazole, por lo que no habría posibilidades de terapia médica combinada.
Una vez que se inicia la terapia médica, se debe revalorar la cortisoluria en el paciente. Si se ha normalizado, se mantendrá con terapia médica mientras hace efecto la radioterapia. Por otro lado, si el paciente no se ha logrado controlar con la terapia médica disponible o si no lo tolera, se debe considerar como última opción, el realizar la adrenalectomía bilateral, para controlar la secreción excesiva de corticosteroides.
La Figura 6 muestra un algoritmo donde se resume el manejo de la enfermedad de Cushing.
Agradecimientos
Por sus valiosos aportes a todos los miembros de la Asociación Costarricense de Endocrinología, Diabetes y Nutrición, que participaron activamente en las diferentes sesiones donde se revisaron y se discutieron estas.
Abstract
The most frequent functioning pituitary adenomas are those who that produce prolactin (prolactinomas), growth hormone (acromegaly) and ACTH (Cushings disease). There are different opinions about the diagnosis and treatment of these diseases. We present here the guidelines for diagnosis and treatment of these tumors made by the Costa Rican Association of Endocrinology, Diabetes and Nutrition.
The main cause of acromegaly is a pituitary tumor that produces growth hormone. The diagnosis is usually made several years after the disease has started because the physical changes occur very slowly. The initial treatment should be surgery and when it fails, medical therapy with octreotide is the main choice. Radiotherapy can be used although it is limited by the long time it takes to start its effect.
If there is hyperprolactinemia, other causes of prolactin elevation should be ruled out. If a prolactinoma is identified, the treatment almost always will be with dopamine agonists. Surgery should be performed only in macroprolactinomas with visual field changes that do not shrink with medical treatment. The first choice for medical treatment will be bromocriptine although there are other agents like cabergoline and quinagolide.
In Cushing´s disease, there are different tests for screening for hypercortisolism, after which a confirmatory test should be performed. Initial treatment should be surgery followed by radiotherapy. In these cases, medical treatment is less effective than in other pituitary tumors.
Referencias
1. Ben-Shlomo, A y Melmed, S. Acromegaly. Endocrinol Metab Clin North Am. 2001; 30:565-83. [ Links ]
2. Chanson, P. Somatostatin Analogs in the Treatment of Acromegaly: the Choice is Now Possible. Eur J Endocrinol. 2000; 143:573-5. [ Links ]
3. Gayle, C. y Sonksen, P. The Presentation of Acromegaly in General Practice. Practitioner. 1999; 243:110-7. [ Links ]
4. Orme SM, McNally RJQ, Cartwright RA, Belchetz PE. Mortality and cancer evidence in acromegaly: a retrospective cohort study. J Clin Endocrinol Metab. 1998; 83:2730-4. [ Links ]
5. Delhougne B, Deneux C, Abs R, Chanson P, Fierens H, Laurent-Puig P, et al.. The prevalence of colonic polyps in acromegaly: a colonoscopic and pathological study in 103 patients. J Clin Endocrinol Metab. 1995; 80:3223- 6. [ Links ]
6. Giustina A, Barkan A, Casanueva FF, Cavagnini F, Frohman L, Ho K, et al. Criteria for Cure of Acromegaly: a Consensus Statement. J Clin Endocrinol Metab. 2000; 85:526-9. [ Links ]
7. Melmed S, Casanueva FF, Cavagnini F, Chanson P, Frohman L, Grossman A, et al. Guidelines for Acromegaly Management. J Clin Endocrinol Metab. 2002; 87:4054-8. [ Links ]
8. Orrego, JJ y Barkan, AL. Pituitary Disorders. Drug Treatment Options. Drugs. 2000; 59:93-106. [ Links ]
9. Biermasz NR, vanDulken H, Roeffman F. Direct postoperative and follow up results of transphenoidal surgery in 19 acromegalic patients pretreated with octreotide compared to those in untreated matched controls. J Clin Endocrinol Metab. 1999; 84:3551-5. [ Links ]
10. Colao A, Cuocolo A, Marzullo P, Nicolai E, Ferone D, Florimonte L, et al. Effects of 1 year treatment with octreotide on cardiac performance in patients treated with acromegaly. J Clin Endocrinol Metab. 1999; 84:17-23. [ Links ]
11. Freda, PU. Somatostatin Analogs in Acromegaly. J Clin Endocrinol Metab. 2002; 87:3534-6. [ Links ]
12. Barrande G, Pittinotungo P, Coste J, Ponvert D, Bertagna X, Luton JP, et al. Hormonal and metabolic effects of radiotherapy in acromegaly: long term results in 128 patients followed in a single center. J Clin Endocrinol Metab. 2000: 85:3779-85. [ Links ]
13. Newman CB, Melmed S, Snyder PJ, Young WF, Boyay LD, Levy R, et al. Safety and efficacy of long term octreotide therapy of acromegaly: Results of a multicenter trial in 103 patients – a clinical research center study. J Clin Endocrinol Metab. 1995; 80:2768-75. [ Links ]
14. Colao A, Ferone D, Marzullo P, DiSamo A, Cerbone G, Samacchiaro F, et al. Effects of different dopaminergic agents in the treatment of acromegaly. J Clin Endocrinol Metab. 1997; 81:518-23. [ Links ]
15. Abs R, Verhelst J, Maiter D, VanAcker K, Nobels F, Coolens JL, et al. Cabergoline in the treatment of acromegaly: Astudy in 64 patients. J Clin Endocrinol Metab. 1998; 83:374-8. [ Links ]
16. Whitman-Elia, GF y Windham, NQ. Galactorrhea May Be Clue to Serious Problems. Patients Deserve a Thorough Workup. Postgrad Med. 2000; 107:165-8. [ Links ]
17. Molitch, ME. Disorders of Prolaction Secretion. Endocrinol Metab Clin North Am. 2001; 30:585-610. [ Links ]
18. Colao A, DiSarno A, Landi MI, Cirillo S, Samacchiaro F, Guiseppina F, et al. Long term and low dose treatment with cabergoline induces macroprolactinoma shrinkage. J Clin Endocrinol Metab. 1999; 82:3574-9. [ Links ]
19. Wilson, CB. Surgical Management of Pituitary Tumors. J Clin Endocrinol Metab. 1997; 82:2381-5. [ Links ]
20. Molitch, ME. Medical Treatment of Prolactinomas. Endocrinol Metab Clin North Am. 1999; 28:143-69. [ Links ]
21. Pinzone JJ, Katznelson L, Danila DC, Pauler DK, Miller CS, Kibanski A. Primary Medical Therapy of Micro and Macroprolactinomas in Men. J Clin Endocrinol Metab. 2000; 85:3053-7. [ Links ]
22. Colao A, DiSarno A, Sarnacchiaro F, Ferone D, DiRenzo G, Merola B, et al. Prolactinomas resistant to standard dopamine-agonists respond to chronic cabergoline treatment. J Clin Endocrinol Metab. 1997; 82:876-83. [ Links ]
23. Tsigos C, Chrousos G. Differential diagnosis and management of Cushing´s syndrome. Annu Rev Med. 1996; 47:443-461. [ Links ]
24. Ross NS. Epidemiology of Cushing´s syndrome and subclinical disease. Endocrinol Metab Clin North Am. 1994; 23:539-46. [ Links ]
25. Morante, Lucas. Síndrome de Cushing. Rev Clin Esp. 1994; 194: 56-68. [ Links ]
26. Kirk L, Hash RB, Katner HP, Jones T. Cushing´s disease: Clinical manifestations and diagnostic evaluation. Am Fam Physician. 2000; 62: 1119-27. [ Links ]
27. Meier CA, Biller BM. Clinical and biochemical evaluation of Cushing´s syndrome. Endocrinol Metab Clin North Am. 1997; 26: 741-62. [ Links ]
28. Hasinski S. Assessment of adrenal glucocorticoid function. Postraduate Medicine. 1998; 104: 61-70. [ Links ]
29. Katznelson L, Bogan JS, Trob JR, Schoenfeld DA, Hedley-White ET, Hsu DW, et al. Biochemical assessment of Cushing´s disease in patients with corticotroph macroadenomas. J Clin Endocrinol Metab. 1998; 83: 1619-23. [ Links ]
30. Tsigos C, Chrousos GP. Physiology of the hypothalamic-pituitary-adrenal axis in health and in psychiatric and other disorders. Endocrinol Metab Clin North Am. 1994; 23:451-466. [ Links ]
31. Papanicolau DA, Yanovski JA, Cutler GB, Chrousos GP, Nieman LK. A single midnight serum cortisol measurement distinguishes Cushing´s syndrome from pseudo-Cushing´s states. J Clin Endocrinol Metab. 1998; 83:1163-67. [ Links ]
32. Yanovski JA, Cutler GB, Chrousos GP, Nieman LK. The dexamethasone-suppresed corticotropin-releasing hormones stimulation test differentiates mild Cushing´s disease from normal physiology. J Clin Endocrinol Metab. 1998; 83:348-52. [ Links ]
33. Becker M, Aron DC. Ectopic ACTH syndrome and CRH mediated Cushing´s syndrome. Endocrinol Metab Clin North Am. 1994; 23:585-606. [ Links ]
34. Flack MR, Oldfield EH, Cutler GB Jr, Zweig MH, Malley JD, Chrousos GP, et al. Urine free cortisol in the high dose dexamethasone suppression test for the differential diagnosis of the Cushing syndrome. Ann Intern Med. 1992; 116:211-217. [ Links ]
35. Klibanski A, Zervas NT. Diagnosis and management of hormone secreting pituitary adenomas. N Engl J Med. 1991; 324:823-31. [ Links ]
36. Nieman LK, Oldfield LH, Wesky R, Chrousos GP, Loriaux DL, Cutler GB Jr. Asimplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of adrenocorticotropin dependent Cushing´s syndrome. J Clin Endocrinol Metab. 1993; 77:1308-12. [ Links ]
37. Oldfield EH, Doppman JL, Nieman LK, Chrousos GP, Meller DL, Kats DA, et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing´s syndrome. N Engl J Med. 1991; 325:897-905. [ Links ]
38. Blevins LS, Christy JH, Khajavi M, Tindall GT. Outcomes of therapy for Cushing´s disease due to adrenocorticotropin-secreting pituitary macroadenomas. J Clin Endocrinol Metab. 1998; 83:63-7. [ Links ]
39. Estrada J, Boronat M, Melgo M, Magallon R, Millan I, Diez S, et al. The long term outcome of pituitary irradiation after unsuccesful transphenoidal surgery in Cushing´s disease. N Engl J Med. 1997; 336:172-7. [ Links ]
Médico cirujano especialista en Endocrinología, servicio de Endocrinología, Hospital San Juan de Dios, Caja Costarricense de Seguro Social. Profesor del Departamento de Farmacología y Toxicología Clínica, Universidad de Costa Rica.
Abreviaturas: IMC, índice de masa corporal; ACEDYN, Asociación Costarricense de Endocrinología, Diabetes y Nutrición; IGF-1, factor de crecimiento tipo insulina-1; TAC, tomografía axial computadorizada; HC, hormona del crecimiento; GHRH, hormona liberadora de hormona de crecimiento; RMN, resonancia magnética nuclear; CBG, globulina transportadora de cortisol.
Correspondencia: Chih Hao Chen Ku, apartado 1406-1200 Pavas, Costa Rica. Fax (506) 207-4489. E-mail: chenku@racsa.co.cr

