








Serviços Personalizados
Journal
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkActa Médica Costarricense
versão On-line ISSN 0001-6002versão impressa ISSN 0001-6012
Acta méd. costarric vol.45 no.2 San José Jun. 2003
Resumen
La enfermedad de alzheimer (EA), es muy frecuente en la vejez donde se pierden, en forma lenta y progresiva, capacidades cognoscitivas y motoras. Esta enfermedad ha ido en aumento conforme se incrementa la expectativa de vida de la población. En los últimos años, descubrir un tratamiento efectivo para esta enfermedad ha sido un verdadero reto científico. A pesar de que su patología fue descrita hace algunos años, no hubo tratamiento disponible hasta hace poco tiempo. Desde principios de los noventa se inició el estudio de los inhibidores de la colinesterasa. Se presentó así, por primera vez, una probable solución sintomática a esta enfermedad. En 1993 salió al mercado la tacrina, primer medicamento de esta clase. En la actualidad hay más variedad de tratamientos, algunos han dado excelentes resultados, como es el caso de la rivastigmina. A pesar de que todos pertenecen a la misma clase farmacológica, hay grandes diferencias en cuanto a su especificidad, su selectividad y efectos secundarios. A continuación se describen y analizan los tratamientos disponibles para esta enfermedad.
Palabras clave: Inhibidores de colinesterasa, enfermedad de Alzheimer, tratamiento, rivastigmina.
Recibido: 07 de enero, 2003
Aceptado: 18 de marzo, 2003
Abreviaturas: EA, enfermedad de alzheimer
Sin embargo, es hasta hace poco que se empieza a obtener resultados tangibles en cuanto a un tratamiento adecuado. Al aumentar la esperanza de vida en la población costarricense, esta enfermedad se convierte en un verdadero problema social, ya que la edad avanzada es uno de los principales factores de riesgo para la EA.
En el próximo cuarto de siglo, la proporción de la población estadounidense de 65 años y más se espera que se incremente en un 50% aproximadamente, lo cual aumentará la frecuencia de enfermedades relacionadas con la edad. 3 Esta es una enfermedad que acarrea problemas serios no sólo en Costa Rica, sino en el ámbito mundial.
La EA es caracterizada por una pérdida progresiva de la memoria y las funciones cognitivas, que afecta a 15 millones de personas alrededor del mundo. La incidencia aumenta un 0.5% por año, después de los 65 años, hasta un 8% por año, después de los 85 años. 4
Al inicio, esta enfermedad no pasó de ser una curiosidad científica. Con el paso de los años se ha convertido en una patología no solo muy frecuente, sino de un impacto tremendo en la salud, por las inconveniencias y sufrimientos que ocasiona a la familia y el costo económico que conlleva. En Estados unidos se calcula el costo de esta patología en 75 billones de dólares por año, con una carga muy estresante para familiares y cuidadores. Por esta razón, debe evaluarse el costo de la enfermedad y el de los medicamentos, que suelen ser caros. 5
La memoria, el aprendizaje y el comportamiento, tan alterados en la EA, componen una sucesión de mecanismos diferenciados, donde cada uno puede depender de procesos psicobiólogicos y neuroquímicos diferentes. 6 Por esta razón el tratamiento debe ir enfocado a los componentes neurobioquímicos del cerebro.
Setenta años después de la descripción de Alzheimer, en Londres, Davis y Maloney descubrieron la falta de un neurotransmisor: la acetilcolina en los cerebros de estos pacientes. 7 Ellos encontraron una marcada disminución de la acetilcolina y dieron nombre a lo que hoy conocemos como: "hipótesis hipocolinérgica" de la enfermedad de Alzheimer. Se ha visto sobre todo en áreas específicas: el hipocampo, cortex frontal, parietal, temporal y todavía más temprano: en el cortex entorrinal, el girus cingulado y otras áreas. 8 Este déficit se ha encontrado también en pacientes con Parkinson, demencia por cuerpos de Lewy, demencia vascular y otras demencias. 2 ,9 ,10 Los síntomas de cualquier tipo de demencia, sin importar la causa, se deben a este déficit colinérgico. Esto produce una falla en la neurotransmisión acetilcolinérgica y degeneración de los circuitos neuronales. 2
Desde luego, la lógica fue: si el déficit en los cuadros demenciales es la acetilcolina, como había sido la dopamina en la Enfermedad de Parkinson, la solución parecía fácil: dar acetilcolina para compensar su déficit. La primera estrategia fue utilizar los precursores de la acetilcolina, como la lecitina o la colina; pero era necesario llegar a dosis muy altas. El efecto no fue lo que se esperaba. Se ideó una nueva estrategia: dar agonistas postsinápticos de la acetilcolina para aumentar el efecto de la poca colina que permanecía en la sinapsis. En este caso, el problema fueron los efectos tóxicos de estas sustancias, por lo que pronto cayeron en desuso.
Luego se pensó en buscar un medio para que la acetilcolina tuviera una vida media mayor. Los inhibidores de la enzima que destruye la acetilcolina parecían el tratamiento ideal, por eso se llaman inhibidores de la colinesterasa.
La acetilcolina está formada por dos componentes, acetato y colina, los cuales se unen mediante la acción de la acetilcolinesterasa. Esta reacción tiene lugar, en su mayor parte, en los terminales nerviosos, más que en otras regiones neuronales. La enzima acetiltransferasa es sintetizada en el cuerpo de la neurona y es transportada mediante flujo axoplásmico a los terminales, donde se activa. La colina es sintetizada en primer lugar en el hígado y es transportada a otros órganos por vía sanguínea. La colina libre se capta específicamente en los terminales nerviosos olinérgicos, mediante una bomba de alta afinidad, dependiente de sodio. La colina está presente en el espacio extracelular, como resultado de la hidrólisis externa de la acetilcolina previamente liberada. El acetato se deriva de la glucosa por la vía del piruvato y del complejo piruvato deshidrogenasa mitocondrial, que genera acetil CoA. La acetilcolina transferasa es una proteína globular que se encuentra en el cerebro. La regulación de la síntesis se debe al hecho de que la bomba de colina de alta afinidad resulta inhibida por un exceso de acetilcolina y acelerada por bajos niveles. 11 (figura 1 ). La acetilcolina es hidrolizada rápidamente (80 a150 microsegundos) por un proceso sucesivo de acetilación de la enzima, separación de la colina y separación del grupo acetilo. 12 Por definición, los inhibidores de la acetilcolinestera intervienen en este proceso, al interactuar con la enzima e inactivarla. De la intensidad con que se fijan a la enzima y de la rapidez con que revierte espontáneamente dicha fijación, dependen la intensidad y la duración de la acción colinesterásica. Las acciones farmacológicas derivan de la inactivación de la acetilcolina, en los sitios donde esta se libere fisiológicamente, tanto en el sistema nervioso central,como en las terminaciones nerviosas periféricas, somáticas o vegetativas. De manera que puede producir efectos adversos, debido a diferentes mecanismos: a) estimulación de los receptores muscarínicos de los órganos efectores vegetativos; b) estimulación, seguida de depresión o parálisis de todos los ganglios vegetativos y de la musculatura esquelética por acción nicotínica; c) estimulación con depresión posterior ocasional, de receptores colinérgicos centrales.6 Algunos de los efectos en el sistema nervioso central son: desincronización del electroencefalograma, activación generalizada y aumento de la situación de vigilia. 13 Este efecto es el que se aprovecha para el tratamiento de EA, donde la actividad colinérgica está disminuida por la pérdida de neuronas de este tipo.
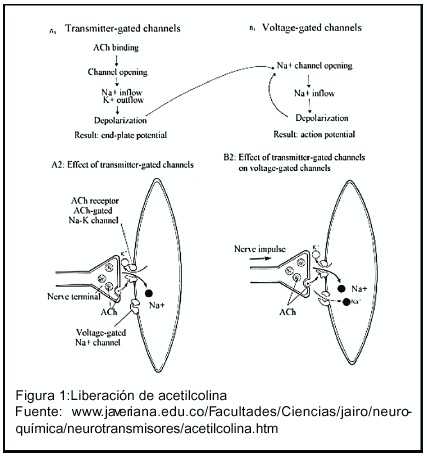
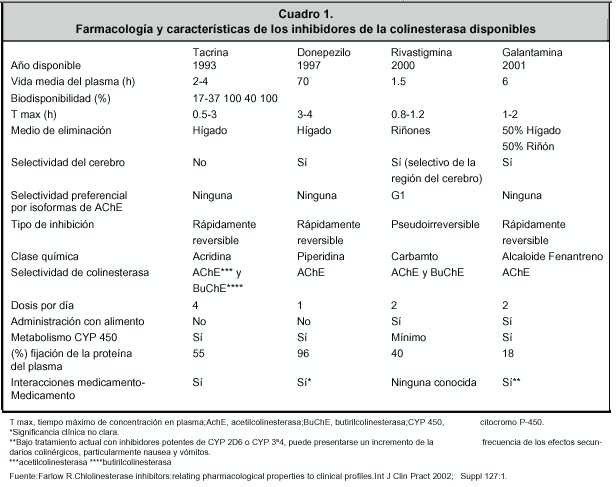
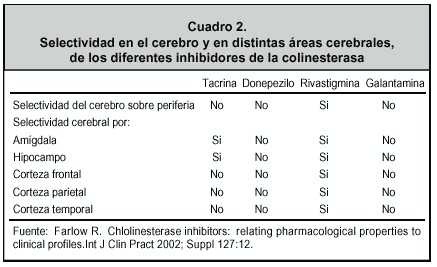
El primero que salió al mercado fue la tetrahidroaminocridina o tacrina que es un inhibidor de acetilcolinesterasa, al igual que de butirilcolinesterasa, de la cual se habla más adelante. No estuvo mucho en el mercado por su hepatotoxicidad. Tanto su efecto en el hígado como, el hecho de que debe tomarse cuatro veces al día, lo hacía impráctico. La acción inhibidora doble: acetilcolina más butirilcolina, hizo a esta sustancia muy atractiva. Luego aparecieron otras sustancias con esta misma ventaja y una mayor afinidad por áreas cerebrales afectadas, por lo cual ahora se usa poco 17 (Cuadro 2 ).
Además, se ha encontrado que la inhibición de la butirilcolinesterasa afecta la formación y la maduración de amiloide y hace que este sea menor o, por lo menos, que las placas inertes no se transforman en dañinas. Al inhibirse la butirilcoli-nesterasa, se evita la transformación de la placa de amiloide en una inflamatoria, lo cual se ha podido deducir del alto contenido de esta en las placas maduras. 22
Igualmente importante es el hallazgo de los isómeros de estas enzimas y su distribución. Las formas G1 aumentan o se mantienen en pacientes con Alzheimer, en relación con las isoformas G2 y G4. Esto puede seguirse de una manera fiel, haciendo estudios de estas isoformas en líquido céfaloraquídeo. 23
En voluntarios sanos con una dosis única de rivastigmina, se logró determinar que la capacidad inhibitoria de butirilcolinesterasa fue del 10% y del 6% para la acetilcolinesterasa en eritrocitos, mientras que en LCR fue de 38.9% para la primera y del 9.7% para la segunda, pero no sucede lo mismo cuando estos estudios se realizan con pacientes que tienen EA, en quienes la inhibición es mayor. 23
Como todos sabemos, las colinesterasas se encuentran en otras áreas diferentes al cerebro, como la sangre, el hígado, el intestino o la unión neuromuscular, pero en el caso de las demencias, nos interesa su acción en las áreas cerebrales.
La tacrina carece de esta afinidad a los tejidos cerebrales, por lo que su acción es equiparable tanto en el cerebro como en los eritrocitos. Esto ocasiona muchos efectos colaterales. Uno de estos es la alteración hepática, por lo que ya no se utiliza. 24
El Donepezilo tiene efectos centrales más específicos, pero también tiene acción sobre la placa neuromuscular. Esto produce algunos efectos indeseables como calambres en las piernas e incontinencia urinaria. Algo similar sucede con la galantamina. Hasta el momento el único fármaco con alta afinidad cerebral es la rivastigmina, lo que debe, en parte, a su acción lipofílica. Recordemos que el cerebro es graso y es ahí donde interactúa con la acetilcolinesterasa y la butirilcolinesterasa. 24 (Cuadro 2 )
La rivastigmina es 5 veces más afín a la forma G1, que justamente es la que aumenta en el cerebro de pacientes con EA. Al contrario de otras formas como la G2, que está más presente en el músculo esquelético y la forma G4, en la unión neuromuscular. 25
También son de interés los efectos de estos medicamentos en largo plazo. Un estudio reciente ha sugerido que el tratamiento crónico con donepezilo produce incremento de los receptores de la colinesterasa, produciendo tolerancia. La rivastigmina, al ser reversible lentamente (pseudo irreversible) (Cuadro 1 ), no produce este efecto, por lo que es posible que sea a largo plazo beneficiosa. 25
La forma G1 es la que aumenta en cerebros de pacientes con Alzheimer. Esto puede explicar por qué los calambres musculares, la incontinencia urinaria o las manifestaciones cardiacas no aparecen con la rivastigmina y sí pueden hacerlo con los otros inhibidores de la colinesterasa. Este isómero G1 aumenta en el hipocampo y en el córtex frontal, áreas muy importantes en la atención.
Se ha visto que en pacientes tratados con rivastigmina por más de 1 mes (3-6 miligramos), se produce un aumento del flujo cerebral en los lóbulos frontal, temporal y parietal del cerebro. El estudio con tomografía de emisión de positrones (Positron Emisión Tomography: PET) ha mostrado una mejoría en el metabolismo de la glucosa en el córtex prefrontal derecho, por más de 12 meses de tratamiento 26 áreas muy importantes en el aprendizaje y la memoria de trabajo y la atención. 27
Este mismo efecto se ha mostrado no solo en el córtex cerebral prefrontal, sino también en el hipocampo, en la amígdala, y es quizá lo que explica el efecto tan claro de este medicamento sobre los trastornos del comportamiento. ( Cuadro 2 ) Estos trastornos, una vez presentes, tienden a progresar si no se da un medicamento que actúe sobre ellos. La depresión, la ansiedad y la agitación son los más frecuentes: la agresión que puede producirse es particularmente difícil de manejar y es quizás la primera causa de institucionalización de estos pacientes. 28
Las ideas paranoides, las alucinaciones muy claras, casi siempre visuales, la incontinencia urinaria, así como los movimientos estereotipados y repetitivos, exasperan hasta a los cuidadores más entrenados y todo obedece a interrupciones en los circuitos que comunican las neuronas, como también sucede en las demencias vasculares y al déficit de los neurotransmisores que ya hemos comentado. 29
La afectación de los lóbulos frontales y temporales, típica en esta patología, explica los trastornos del comportamiento y los cambios emotivos. La causa subyacente de estas alteraciones es un déficit colinérgico en el área límbica o paralímbica, por eso responden a las sustancias que mejoran ese neurotransmisor. Esta es la razón por la cual las drogas que vayan a modificar positivamente estos trastornos del comportamiento, van a ser muy apreciadas.
Desde luego que los antipsicóticos, conocidos como inhibidores de la dopamina, disminuyen la agresión, la agitación y la psicosis. El inconveniente de estos fármacos se relacionan con efectos colaterales, como síntomas extrapiramidales muy molestos, especialmente en la enfermedad por cuerpos de Lewy, en la demencia del Parkinson, y pueden incrementar el déficit cognitivo preexistente. La rivastigmina afecta posi-tivamente estos cambios, al aumentar la vida del neurotransmisor que hace falta para mejorar la memoria. 29
Además, la rivastigmina se elimina por vía renal. Esta es una gran ventaja, porque en el caso de necesitarse un antipsicótico, este puede utilizarse sin problema, dado que, la rivastigmina no emplea la vía del citocromo P 450, como sí sucede con otros inhibidores de la acetilcolinesterasa (Cuadro 1 ).
Hay estudios en los cuales se combina la utilización de rivastigmina con la risperidona. 30 Sin embargo, todavía no hay resultados concluyentes sobre esta combinación.
Estos medicamentos no van a ser la cura de la enfermedad, pero sí puede influenciar positivamente la clínica de la patología: en el ámbito cognitivo, del comportamiento y de actividades de la vida diaria. Todo esto va a permitir una mejor calidad de vida para el paciente y los cuidadores, y en todo caso, retrasar la institucionalización.
Se ha repetido muchas veces, que el tratamiento de los síndromes demenciales debe iniciarse con le explicación del diagnóstico a los familiares. Se debe plantear el horizonte de cómo va a evolucionar la enfermedad y conseguir, que en todo caso, los cuidadores sientan que pueden contar con apoyo. A lo largo del tiempo se puede decir que el mayor enemigo es el miedo al futuro, a sentirse solo, a que nadie ayude cuando se necesite. Este conocimiento va a propiciar una mejor relación entre toda la familia y evitar roces.
También es importante que el médico ayude en los altos y bajos de la enfermedad, en estabilizar las patologías concurrentes o subyacentes, ya evitar la sobremedicación. Se debe colaborar con a la familia para eliminar gastos que no son necesarios; se trata de una enfermedad cuya evolución es larga (más o menos 12 años), de modo que la fármaco-economía cuenta mucho. Se debe seguir al paciente y evitar hacer un diagnóstico y dejar a los familiares a la deriva; es necesario que el médico acompañe y guíe en todo el proceso.
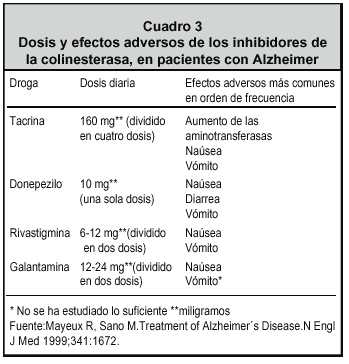
La galantamina se debe iniciar con 4 miligramos dos veces al día y luego se suben 4 miligramos cada 15 días hasta llegar a una dosis de 12 miligramos, luego 16 y raramente 24 miligramos, haciendo la escalada mensual. 2
La rivastigmina se inicia con 1.5 miligramos dos veces al día, a veces 1.5 miligramos por la noche, luego se va subiendo la dosis de forma muy suave. Uno de los grandes problemas es aumentar la dosis muy rápidamente, dado que los efectos colaterales estropean una buena oportunidad para el paciente. Otro problema consiste en no esperar el tiempo necesario para que los fármacos actúen en la dosis recomendada; este lapso debe ser por lo menos dos meses.
Los trastornos del sueño son un acompañante de las demencias tipo alzheimer, Parkinson y por cuerpos de Lewy. El donepezilo reduce la latencia de la etapa del sueño MOR (movimientos oculares rápidos), por lo que hay quienes no lo usan en personas con problemas importantes del sueño, lo que no sucede con la rivastigmina. 33 En cuanto a los efectos cardiovasculares bradicardia y extrasistolia) que se presentan a veces y que no suelen ser muy graves, no se han reportado con la rivastigmina; lo mismo se puede decir de los calambres en las piernas o la incontinencia urinaria. En un análisis de cuatro estudios de casos y controles en Enfermedad de Alzheimer se demostró que no hay asociación entre el tratamiento con rivastigmina y cambios en la frecuencia cardiaca, ritmo, presión arterial, anormalidades de electrocardiograma o frecuencia respiratoria. 34 Ya se había mencionado que la interacción con otras drogas es muy poco probable. La rivastigmina no utiliza la ruta del citocromo para su eliminación. Esto es particularmente importante de tener en cuenta, ya que las personas mayores poseen múltiples patologías y reciben muchos fármacos a la vez. En gerofarmacología, cada día se tiende a usar medicamentos con pocas interacciones, o por lo menos con las menores.
En el horizonte hay otras formas de tratamiento, como la vacuna contra el Alzheimer, propugnada en los laboratorios Elan, con el Dr. Schenk; era una excelente propuesta, sin embargo se presentaron 15 casos de meningoencefalitis, por lo que se detuvo su uso. En este momento hay otra forma de vacunación que se está estudiando en Suiza, el Dr. Hoeck, pero es muy temprano para tener conclusiones. 35
El Dr. Dennos Selkoe, de la universidad de Harvard, sigue encabezando un grupo de investigadores que está tratando de evitar la formación del amiloide o lograr que, por lo menos, este sea más soluble con un grupo de enzimas llamadas se-cretasas. Hay promesas con la gamma secretasa, pero no son más que eso. 3
Otro campo que tuvo y aún tiene muchos adeptos es el de los antinflamatorios. Las citokinas cada vez tienen más importancia en esta patología. También se podrían citar los antioxidantes y los estrógenos, pero no existe base científica para su uso. 3
Abstract
Referencias
1. www.ciudadfutura.com/temassalud/articulos/alzheiach.pdf [ Links ]
2. Farlow RM. Chlolinesterase inhibitors: relating pharmacological properties to clinical profiles. Int J Clin Pract 2002; Suppl 127: 1-5. [ Links ]
3. Zurad E. Nuevos tratamientos de la Enfermedad de Alzheimer:una revisión. Drug Benedit Trends 2001: 1-9. [ Links ]
4. Makeup R, Sano M. Treatment of Alzheimer’s disease. N Engl J Med 1999; 341: 1671-9 [ Links ]
5. Frolich L. Psychosocial care in Alzheimer’s disease:a clinician’s perspective. Alzheimer Insights 1999; Jan:18-20. [ Links ]
6. Flórez J. Farmacología humana. España:Editorial Masson, 1997:222-224, 594-596. [ Links ]
7. Davies K, Maloney, A. Selective loss of central cholinergic neurons in Alzheimer disease. Lancet 1976; 2: 1403-7. [ Links ]
8. Geula C, Mesulam M-M. Chonigergic systems and related neropathological predilection patterns in Alzheimer Disease. In Terry RD, Katzman R, Bick KL, Eds. Alzheimer Disease. New York, USA: Raven Press Ltd, 1994: 263-291. [ Links ]
9. McKeith I. Dementia with Lewy bodies. Br J Psychiatry 2002; 180: 144-147. [ Links ]
10. Esiri MM, McShane RH. Parkinson’s Disease and Dementia. In: Esiri MM, Morris JH, Eds. The neuropathology of dementia. Cambridge, UK: Cambridge University Press, 1997; Chapter 6: 174-193. [ Links ]
11. www.javeriana.edu.co/facultades/ciencias/jairo/neurobioquimica/neurotransmisores/acetilcolina.htm [ Links ]
12. Arendt T. Mecanismos colinérgicos de trastornos del comportamiento en Enfermedad de Alzheimer. Alzheimer Insights 2001; Feb:1-3. [ Links ]
13. www.medmayor.cl/apuntes/farmacologia/colinergicos,%20adrenergicos%20y%20serotoninergicos.htm . [ Links ]
14. Greig N et al. Butirilcolinesterasa: un nuevo objetivo terapéutico en el tratamiento de la Enfermedad de Alzheimer? Alzheimer Insights 2001; Jun 1-3. [ Links ]
15. Bryant J et al. Clinical and cost-effectiveness of donepezil, rivastigmine and galantamine for Alzheimer Disease: a rapid and systematic review. Health Technol Assess 2001; 5:1-137. [ Links ]
16. Doody R et al. Practice Parameter: Managemente of dementia (an evidence based review). Reports of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001; 56: 1154-1166. [ Links ]
17. Gray A. Meeting the economic challenges of Alzheimer’s disease: will drug interventions ease the burden? Alzheimer Insights 2000; May: 4-6. [ Links ]
18. Farlow M et al. A 52-week study of the efficacy of rivastigmime in patients with mild to moderately severe Alzheimer’s disease. Eur Neurol 2000; 44: 234-241. [ Links ]
19. Mesulam M et al. Widely spread butyrylcolinesterase can hydrolyze aceticholine in the normal and Alzheimer brain. Neurobiol Dis 2002; 9:88-93. [ Links ]
20. Lie B et al. Abundant tissue butyrylcolinestyerase and its posible function in the acetylcholinesterase knockout mouse. J Neurochem 2000; 75: 1320-1331 [ Links ]
21. Roesler M, Retz W, Retz Junginger P, Dennler HJ. Effects of two-year treatment with the cholinesterase inhibitor rivastigmine on behavioral symptoms in Alzheimer´s Disease. Behav Neurol 1998; 11: 211-216. [ Links ]
22. Arendt T. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer. Neurochem1992; 4:12-17. [ Links ]
23. Poirier J. Evidence that the clinical effects of cholinesterase inhibitors are related to potency and targeting of action. Int J Clin Pract 2002; Suppl 127: 6-17. [ Links ]
24. Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract 2002; Suppl 127; 45-58 [ Links ]
25. Davidsson P et al. Differential Increase in cerebrospinal fluid-acetylco-linesterase after treatment with acetylcolinesterase inhibitors in patients with Alzheimer Disease. Neurosci Lett 2001; 300: 157-160. [ Links ]
26. Venneri A et al. Cerebral blood flow and cognitive responses to rivastigmine treatment in Alzheimer disease. Neuro Report 2002; 5:23-27. [ Links ]
27. Nordberg A et al. Improved cortical glucose metabolism in Alzheimer with patients treated with rivastigmine for one year. J Neurol Sc 2001; 187: 14-18. [ Links ]
28. Doncel B, Drachman D. Incontinent and troublesome behaviors predict institutionalizations. J Geriatric Psychiat 1992; 3: 12-16. [ Links ]
29. Del Ser T. Dementia with Lewy Bodies. Alzheimer Insights 2001; May: 1-3. [ Links ]
30. Weiser M, Rotmensch H. Coadministration of rivastigmine with risperidone in dementia patients with behavioral disturbances. Int. J. Geriat. Psychiat 2002; 6:28-32. [ Links ]
31. Corey-Bloom J, Anad R, Messina J, Veach J, for the ENA-713 B352 Study Group. A randomized trial evaluating the efficacy and safety of ENA-731 (rivastigmine tartrate),a new acetylcolinesterase inhibitor, in patients with mild to moderately severe Alzheimer´s Disease. Int J Geriatr Psychofarmacol 1998; 1:55-65. [ Links ]
32. Rosler M et al. Efficacy and safety of rivastigmine in patients with Alzheimer´s Disease: international randomized control trial. BMJ 1999; 318: 633-638. [ Links ]
33. Schredl M et al. Donepezil-induced REM sleep augmentation,enhances memory performance in elderly, healthy persons. Exp Gerontol 2001; 36: 353-361. [ Links ]
34. Potkin S, Messina J, Graham S. Safety of rivastigmine in Alzheimer´s Disease. Neurol 2000; 54: 2261-2268. [ Links ]
35. McIntosh A. Manejo en equipo de la demencia en cuidado primario. Visión del R.U. Alzheimer Insights 2001; 7:8-11. [ Links ]
1 Médico especialista en Geriatría y Gerontología, área de interés envejecimiento cerebral.
2 Médico General. Graduado de la Universidad de Costa Rica.
Correspondencia: Carlos Luis Alpízar-Quesada. Correo electrónico: calpizar@hospitalsanjose.net .
Catalina Morales-Alpízar. Correo electrónico: catamoralesalpizar@hotmail.com .