










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.42 n.1 San José Mar. 2000
Resumen:
El objetivo de este estudio fue identificar cromosomopatía fetal en voluntarias con embarazos de alto riesgo genético, a fin de brindar adecuada atención obstétrica y pediátrica y asesoramiento genético. Las células fetales se obtuvieron mediante amniocentesis (N=506) y cordocentesis (N=46) desde 1993 hasta 1998 inclusive. Ambas punciones fueron transabdominales, guiadas por ultrasonografía y se realizaron en los hospitales Calderón Guardia (63% de las amniocentesis y 45 cordocentesis), México (21% de las amniocentesis y una cordocentesis), en la consulta privada (12%) y otros hospitales. La indicación del 62% de las amniocentesis y de casi todas las cordocentesis fue el examen ultrasonográfico anormal y el 23% de las punciones fue por edad materna avanzada. El 66% de las veces el estudio se realizó en la segunda mitad del embarazo. De las 552 muestras de líquido amniótico y sangre fetal, en 109 no fue posible obtener resultados. Los 443 cariotipos fetales obtenidos fueron anormales en 39 casos (9%): 21 cariotipos trisómicos, ocho casos con síndrome de Turner (45,X), tres mosaicos cromosómicos y siete cariotipos anormales por otras causas. El resultado final se obtuvo en 15 días (mediana). En el seguimiento de los casos se encontró concordancia entre el cariotipo y el fenotipo del recién nacido, al igual que entre el diagnóstico ultrasonográfico fetal y la condición del neonato. El diagnóstico prenatal de cromosomopatía permitió el asesoramiento genético y el manejo obstétrico y pediátrico de los casos de manera adecuada. En los embarazos con cariotipo normal, esta información alivió la preocupación de muchos de los padres.
Descriptores
Diagnóstico prenatal, amniocentesis genética, embarazo de alto riesgo, cromosomas fetales, anomalías intrauterinas.
Las malformaciones congénitas y las complicaciones respiratorias asociadas a prematuridad son las principales causas de la mortalidad infantil en Costa Rica.1 Las aberraciones cromosómicas son responsables de una parte importante de los defectos congénitos, por lo que la prevención de las mismas es una tarea de fundamental importancia. La prevención primaria de cromosomopatía fetal se dificulta por el hecho de que la mayoría de los defectos cromosómicos no son de origen hereditario sino más bien producto de mutaciones nuevas, imposibles de anticipar,2 de modo que la prevención primaria se limita a identificar factores de riesgo conocidos de cromosomopatía. Con este enfoque, es poca la disminución que se puede lograr en la morbimortalidad perinatal por defectos cromosómicos. Las cromosomopatías afectan 38-75/1000 embriones en el I trimestre, 27/1000 fetos de 15 a 20 semanas, 6.5/1000 recién nacidos y 5/1000 niños entre 7-8 años de edad.3 En los fetos muertos de más de 28 semanas gestacionales, el 12% de los macerados tienen aberraciones cromosómicas y el 4% de los no macerados. De los bebés que mueren en las 4 primeras semanas de vida extrauterina, el 6% posee cromosomopatía.3 En los sobrevivientes, la mayoría de estos defectos son invalidantes, producen individuos polimalformados y con retardo mental, en los cuales las posibilidades de la prevención terciaria son también limitadas. De esta manera, la prevención secundaria, una vez ocurrida la patología o post-concepción, es la mejor alternativa de manejo de esta situación.4 La meta es el diagnóstico de patología fetal lo más temprano en el embarazo para valorar la posibilidad de tratamiento intrauterino, interrupción del embarazo, o preparación del núcleo familiar y del personal de salud, para la atención óptima del neonato afectado, y así minimizar el daño y optimizar el tratamiento o rehabilitación.
El diagnóstico cromosómico fetal mediante amniocentesis y cultivo de las células fetales descamadas en el líquido amniótico, es el método más utilizado, forma parte de las normas de atención de la mujer embarazada de alto riesgo en la mayoría del mundo desarrollado5,6 y es un componente indispensable de los programas preventivos de genética que impulsa la Organización Mundial de la Salud.4
La experiencia del diagnóstico fetal citogenético en Costa Rica, a propósito de los primeros 186 casos estudiados de 1985 a 1992, mostró que la amniocentesis para detectar anomalías cromosómicas es un método seguro y confiable.7
El objetivo de este estudio fue identificar cromosomopatía fetal en voluntarias con embarazos de alto riesgo genético, a fin de brindar adecuada atención obstétrica y pediátrica y proporcionar asesoramiento genético.
Materiales y Métodos:
Este estudio prospectivo se basó en la captación, sin método de muestreo, de 552 embarazadas con alto riesgo de cromosomopatía fetal desde enero de 1993 hasta diciembre de 1998. La captación de los casos ocurrió en la consulta prenatal y en las unidades de perinatología de los hospitales R.A. Calderón Guardia (N=361), México (N=115), San Juan de Dios (N=2), Hospital Materno Infantil Carit (N=8), Hospital Max Peralta (N=2) y en la consulta privada (N=64).
Previo consentimiento informado, 506 pacientes se some-tieron voluntariamente a una amniocentesis transabdominal, guiada por ultrasonografía, según las técnicas usuales.8 Con igual metodología se realizaron 45 cordocentesis en el H.R.A Calderón Guardia y una más en el H. México. La sangre fetal así obtenida se procesó para análisis citogenético de la manera usual.9
El 94% de las veces la penetración de la cavidad uterina fue única, se repitió en el 6% restante. El calibre de la aguja fue Nº22 en el 81% de las punciones, N°20 en el 15%, N°19 en tres casos y N°18 en siete oportunidades.
La apariencia del líquido amniótico fue normal (51%), turbia (43%), color café (2%) y sanguinolenta (5%).
Los procedimientos de cultivo celular y análisis cromosómico ya han sido descritos,7 la única variación introducida en este estudio es que las muestras del Hospital México fueron de menor volumen, 20 ml de líquido en dos tubos, que por lo tanto fueron procesados por duplicado. Se utilizó el sistema cerrado de histocultivo y la cosecha mediante suspensión. Las preparaciones cromosómicas se bandearon (GTG) y analizaron utilizando la nomenclatura y recomendaciones internacionales.10
Tomando en cuenta que las dos o tres botellas de cultivo no siempre están listas para cosecha simultáneamente, el total de días hasta obtener el resultado final osciló entre 7 y 58, el promedio fue 17 días, la mediana 15 y la moda 14 días; el 55% de los diagnósticos se tardó entre 7 y 15 días, a los 22 días se obtuvo el 84% de los resultados y el 95% al mes.
Resultados:
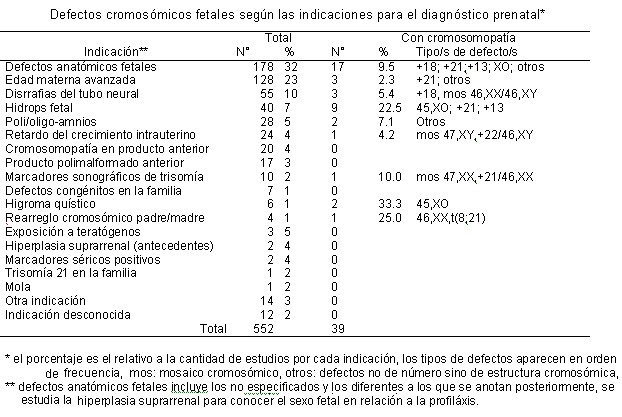
Las indicaciones para realizar las amniocentesis y cordocentesis se muestran en el Cuadro 1. Las amniocentesis se realizaron en las siguientes edades gestacionales: semanas 10 a 16 (N=37), semanas 17 y 18 (N=93), semanas 19 y 20 (N=52), semanas 21 a 27 (N=117) y 28 o más semanas (N=194). En 13 casos no se informó la edad gestacional. Las edades gestacionales en que se realizaron las cordocentesis fueron 18 semanas (N=1), de 21 a 27 semanas (N=12) y de 28 o más semanas (N=33).
En 106 de las 506 muestras de líquido amniótico no fue posible obtener el cariotipo fetal por: ausencia de crecimiento celular (N=72), la cosecha rindió preparaciones cromosómicas de calidad o cantidad insuficientes (N=21), contaminación bacteriana poco después de iniciado el cultivo y antes de efectuar el primer cambio de medio de cultivo, por lo que pudieron con-taminarse al momento de tomar las muestras (N=5), contaminación luego de uno o varios cambios de medio de cultivo (N=4) y combinaciones de varias causas de fracaso (N=4). Respecto a las muestras que fracasaron en crecer y la apariencia del líquido, el 12% de los casos de apariencia normal no crecieron, lo mismo que el 16% de los casos de líquido turbio, el 32% de las muestras sanguinolentas no crecieron y tampoco creció el 22% de los líquidos color café. La edad gestacional de la muestra fue otro factor que influyó en el fracaso en el crecimiento celular, el 70% de las muestras que no prosperaron eran de edades gestacionales superiores a las 20 semanas. En el caso de las 46 cordocentesis, cuatro muestras no crecieron por venir diluídas con líquido amniótico.
Los 443 cariotipos fetales obtenidos fueron femeninos normales en 197 estudios, masculinos normales en 207 casos y anormales en 39 casos (Cuadro 2). Además se detectaron dos pseudomosaicos: 46,XX,t(1;2)(q21;q31)/46,XX y 46,XX,inv(17)/46,XX en células provenientes del líquido amniótico. En una ocasión en que se cultivaron líquido amniótico y sangre obtenida por cordocentesis, de un mismo caso, el cariotipo en la muestra de líquido fue masculino y el de la muestra de sangre fue femenino, lo que hace suponer que la sangre no era fetal sino materna. Por esta misma razón, es posible explicar la presencia de células XX y células XY en una muestra obtenida por cordocentesis (Cuadro 2).
| Cariotipo | | |
| __________________________________________________________ | ||
| No se obtuvo Normal: | | |
| 46, XX | | |
| | | |
| Trisomías | ||
| 47, + 21 | | |
| 47, + 18 | | |
| 47, + 13 | | |
| Síndrome de Turner: | | |
| 45, X | | |
| Mosaicos: | | |
| 47, XY, + 22/46, XY | ||
| 47, XX, + 21/46, XX* | ||
| 46, XX / 46, XY* | ||
| Defectos estructurales: | | |
| 46, XY,der (3)t(3;9)pat | ||
| 46, XY,der (15) | ||
| 46, XY, der (4) | ||
| 46, XX, ins(16;?)(q12;?) | ||
| 46, XX,t(8;21)mat | ||
| Triploidía: | | |
| 69, XXX | ||
| Cromosoma extra estructuralmente anormal: | | |
| 47, XY, + inv dup (15)pat | ||
| _________________________________________________________ | ||
| Total | | |
| _________________________________________________________ | ||
El seguimiento de los casos se realizó, hasta el año 1996, para 95 de las pacientes estudiadas. Se encontró, dentro de la primera semana post-punción, contracciones uterinas de moderada intensidad (N=4), labor prematura (N=5), sangrado vaginal (N=1), ruptura prematura de membranas (N=1) y muerte fetal de un producto polimalformado (N=1). En 83/95 (87%) embarazadas no se presentó ninguna complicación. De una semana post-punción en adelante se produjeron cuatro abortos y 90 partos, la mitad de los cuales fue mediante operación cesárea. Las indicaciones de cesárea fueron hernias medulares, gastrosquisis y otras lesiones abiertas (N=11), sufrimiento fetal (N=6), desproporción céfalo-pélvica (N=5), primigesta añosa o edad materna avanzada (N=6), hidrocefalia (N=2), feto hidrópico (N=2), oligoamnios (N=2), masa pélvica (N=2), cesárea anterior (N=2) y un caso cada uno de presentación pélvica, placenta previa, desprendimiento prematuro de placenta, y para extraer tumor de ovario.
En los recién nacidos se encontró concordancia entre el ca-riotipo y el fenotipo, lo mismo que entre el diagnóstico ultrasonográfico fetal y la condición del neonato. En ninguno de estos niños se detectó secuelas de la punción.
Discusión
Estos resultados no son comparables con la mayoría de los informes de casuísticas mundiales, ya que por lo general, las indicaciones de amniocentesis y la edad gestacional a la que se practicaron son diferentes. La mayoría de los centros de diagnóstico prenatal condicionan el estudio a la edad gestacional en que la interrupción del embarazo por defecto fetal es legalmente permitida, de manera que se proponen conseguir el resultado antes de la vigésimocuarta semana de gestación. Como consecuencia, la indicación de amniocentesis más del 80% de las veces fue edad materna avanzada y la frecuencia de cromosomopatía fetal fue alrededor de 5% o menos.6 Por el contrario, en la segunda mitad del embarazo, la principal indicación de amniocentesis, cordocentesis o biopsia placentaria fue examen ultrasonográfico anormal, usualmente por retardo del crecimiento fetal, poli u oligohidramnios, feto hidrópico y malformaciones fetales.11,12 En estas circunstancias cabe esperar frecuencias mayores de ca-riotipos defectuosos, tales como 11%,13 13%,12 14%14 y 23%.15 Esto explica el hallazgo de 9% de cromosomopatía encontrada en este estudio, puesto que la indicación del 62% de las amniocentesis y de casi todas las cordocentesis fue el estudio ultrasonográfico anormal (Cuadro 1) y el 66% de las veces las amniocentesis se realizaron en la segunda mitad del embarazo.
Además de cuantitativas, las diferencias entre una población estudiada primordialmente por edad materna avanzada y otra con predominio de anomalías fetales, son también cualitativas. En 6515 estudios prenatales en mujeres mayores que 38 años,12 la trisomía 21 fue responsable del 1,5% de los cariotipos anormales, la trisomía 18 del 0,3% y la trisomía 13 del 0,1%; mientras que la frecuencia de estos defectos en 936 estudios por ultrasonido anormal12 fue de 2,9% (+21), 3,5% (+18) y 2% (+13). Otros informes apoyan estos mismos ha-llazgos.14 Esta puede ser la causa de la abundancia relativa de las trisomías 18 y 13 encontradas en este estudio (Cuadro 2).
Los pseudomosaicos encontrados, son anomalías que surgen in vitro y que por lo tanto no reflejan la condición fetal. Este fenómeno se presenta en el 0,9% de los cultivos en la expe-riencia mundial.3 Se han establecido pautas bien definidas que permiten diferenciar el pseudomosaico del verdadero ca-riotipo fetal,3,10 las cuales seguimos.
Actualmente se utilizan marcadores sonográficos que permiten, aún a edades gestacionales relativamente tempranas, sospechar la presencia de cromosomopatía fetal.16-18 Estos marcadores son: cabeza "en forma de fresa", quistes del plexo coroides, retardo del crecimiento intrauterino, edema o en-grosamiento de la nuca, acortamiento de los huesos largos, pieloectasia, intestino ecogénico, higroma quístico y malformaciones fetales en general. En los casos de trisomía 18 detectados en este estudio (Cuadro 1), se encontraron tanto malformaciones múltiples como aisladas (mielomeningocele). El hallazgo de espina bífida se asocia a defectos cromosómicos en el 17% de los casos y las anomalías más frecuentes son trisomía 18, trisomía 13, triploidía y translocaciones.19 Los hallazgos sonográficos más frecuentes de los fetos con trisomía 18, de 24 semanas gestacionales o menos, son: higroma quístico, engrosamiento de la nuca y mielo-meningocele.20 Después de la semana 24 de gestación, son más frecuentes el retardo del crecimiento intrauterino, malformaciones cardíacas, y cisterna magna aumentada de tamaño. Los quistes de los plexos coroides se pueden presentar en cualquier edad gestacional.20 En cuanto al higroma quístico de la nuca, de los seis casos estudiados, dos fueron positivos por síndrome de Turner (Cuadro 1), lo cual correlaciona con los informes que asocian esta patología a monosomía X.21
Las trisomías 13 y 18 en recién nacidos son de mal pronóstico, la sobrevida promedio de la trisomía 18 es de cuatro días, el 45% sobreviven hasta una semana, el 3-9% sobreviven hasta seis meses y 0-5% alcanzan el año de edad.22 Los sobrevivientes tienen un promedio de dos operaciones al cumplir su primer año y se desenvuelven en los ámbitos de retardo severo y profundo.23,24
La trisomía 22 en mosaico es un fenómeno raro en el diagnóstico prenatal, se han documentado por lo menos nueve casos en la literatura, además del nuestro (Cuadro 1), en los que el retardo del crecimiento intrauterino también fue un hallazgo frecuente.3
La triploidía (69 cromosomas) se encuentra en alrededor del 12% de los abortos espontáneos del primer trimestre pero es rara en nacidos vivos. Las complicaciones obstétricas de las triploidías completas en el 92% de los casos son: poli u oligohidramnios, retardo del crecimiento intrauterino, degene-ración hidatidiforme de la placenta, preeclampsia y prematuridad. El 100% mueren durante el primer año de vida.25 En el caso nuestro (Cuadro 2), se presentó oligoamnios severo, hidrocefalia y edad materna de 41 años.
Aproximadamente 1:500 nacidos vivos es portador de un rearreglo balanceado de la estructura cromosómica,3 lo cual no afecta su salud pero sí reduce su posibilidad de tener descendencia cromosómicamente normal. Esta es la situación del padre portador de una translocación entre los cromosomas 3 y 9 (Cuadro 2) y de la madre portadora de otra translocación, esta vez entre los cromosomas 8 y 21, cuya hija heredó su mismo rearreglo cromosómico (Cuadro 1). En el primer caso se produjo un cariotipo fetal desbalanceado por trisomía parcial del cromosoma 9 y monosomía parcial del cromosoma 3 y en el segundo caso la niña es portadora sana con riesgo elevado de cromosomopatía en su descendencia, lo mismo que su madre.
Los cromosomas extra, estructuralmente anormales, también llamados marcadores, supernumerarios o accesorios, tienen una prevalencia al nacimiento estimada de 0.14 a 0.72 por mil. Aproximadamente la mitad de las veces se trata de una inversión y duplicación de un segmento, de tamaño variable, del cromosoma 15. El caso nuestro (Cuadro 2), se trató de una duplicación invertida del cromosoma 15 de origen paterno, que se presentó como un pequeño marcador dicéntrico y con doble juego de satélites. En este caso, no cabe esperar consecuencias fenotípicas, tanto por lo pequeño del marcador, que incluye solamente material adicional confinado a q11, sino porque no las presenta el padre del feto.
La cordocentesis que arrojó un cariotipo femenino, mientras que el líquido cultivó células masculinas, lo mismo que uno o ambos mosaicos (Cuadro 2) detectados en muestras obtenidas mediante cordocentesis (la trisomía 21 en mosaico se presenta en alrededor del 1 al 3% de los casos), ilustra la necesidad de verificar la procedencia de la muestra de sangre. Se pueden utilizar marcadores hematológicos (por ejemplo en el feto predomina el linfocito y en la madre el neutrófilo) o bioquímicos, la prueba de Kleihauer-Bekte, determinación del grupo sanguíneo, detección del antígeno I con anticuerpos monoclonales y el mejor marcador para la detección de contaminación de la muestra con sangre materna: concentración de la subunidad ß de la gonadotrofina coriónica humana.26
La alta frecuencia de anomalías cromosómicas en presencia de defectos estructurales fetales y la alta mortalidad fetal, enfatizan la necesidad de obtener el cariotipo. En presencia de cromosomopatía se puede evitar cirugía fetal innecesaria, como sucedió en uno de los casos de trisomía 13 con uropatía obstructiva, que se preparaba para reparación quirúrgica in utero en caso de cariotipo normal. Por otro lado, el conoci-miento del cariotipo y del fenotipo fetal permite a los padres y al personal de salud discutir alternativas y escoger el mo-mento, el modo y el lugar más adecuados para el nacimiento. Aún más, dada la alta probabilidad de muerte intrauterina y maceración fetal consiguiente, el diagnóstico citogenético post-parto resulta poco práctico y por lo tanto el asesora-miento genético a fin de evitar recurrencia se dificulta.
En Costa Rica, con referencia al diagnóstico prenatal, tenemos dos problemas fundamentales: 1) la capacidad del único laboratorio en el país que realiza cariotipos fetales es por motivos económicos restringida y definitivamente insuficiente para atender a toda la población de embarazadas con indicación para amniocentesis genética, 2) las trabas legales para interrumpir embarazos por defecto fetal. Es así como surge una situación de injusticia: en el nivel privado se ofrece el diagnóstico prenatal a las embarazadas mayores que 35 años, asumiendo su capacidad económica para el aborto selectivo fuera de nuestras fronteras. A las aseguradas, se les realiza el estudio fundamentalmente cuando el examen ultrasonográfico es anormal para determinar la posible etiología cromosómica del problema y ofrecer asesoramiento genético con miras a futuros embarazos.
Puesto que está demostrado que los niños con síndrome de Down en quienes se invierte en aceptación, amor, cuidados, estimulación temprana, terapia de lenguaje, terapia ocupacional, fisioterapia, integración en el sistema escolar, etc., pueden llegar a ser estrellas de televisión, resulta paradójico que las clases sociales con menos recursos económicos para costear todas estas terapias e intervenciones y con mayor cantidad de factores de riesgo asociados, sean las que menos posibilidades de prevención, mediante aborto selectivo, poseen.
Abstract
The results of 506 genetic amniocentesis and 46 percutaneous umbilical blood samplings, from 1993 to 1998, are reported. There were two main reasons for referral: abnormal ultrasound assessment (62% of cases) and advanced maternal age (23%). Most procedures (66%) were performed during the second half of pregnancy. Fetal cells were closed cultured and mass harvested. In 9% of cases fetal chromosomes were abnormal, due to trisomies 18, 21 and 13, monosomy X, mosaic trisomies 21 and 22, balanced and unbalanced translocations, extra structurally abnormal chromosomes and other defects. Two cases of pseudomosaicism were detected. Turn around time was 15 days median. Prenatal cytogenetic and sonographic findings correlated with the phenotype of the newborn. Prenatal diagnosis of fetal defects allowed genetic counseling as well as better obstetric management and pediatric care. Normal results of both tests provided reassurance to prospective parents.
Key Words
Amniocentesis, prenatal diagnosis, fetal chromosome analysis, pregnancy, sonography.
Agradecimiento
Este trabajo es financiado por la Vicerrectoría de Investi-gación de la Universidad de Costa Rica, a través de los pro-yectos N°742-84-115 y N°742-95-307 y por la Vicerrectoría de Acción Social de la U.C.R., mediante las resoluciones ED-060-97 y ED-061-97.
Referencias
1. Departamento de Estadística, Sección Otros Programas Prioritarios, Ministerio de Salud. Diagnóstico perinatal por cantón y causa, Costa Rica 1992-1994. San José: Ministerio de Salud, 1995. [ Links ]2. Organización Panamericana de la Salud. Prevención y control de las enfermedades genéticas y los defectos congénitos. Informe de un grupo de consulta. Publicación Científica Nº460. Washington, D.C.: OPS, 1984.
3. Hsu LYF. Prenatal diagnosis of chromosomal abnormalities through amniocentesis. En: Milunsky A, ed. Genetic disorders and the fetus, diagnosis, prevention and treatment. 3a.ed., Baltimore: Johns Hopkins Univ. Press, 1992:155-210. [ Links ]
4. Organización Panamericana de la Salud. Ejecución de las actividades de salud de genética en América Latina y el Caribe. Washington, D.C.: OPS, 1987. [ Links ]
5. Annas GJ, Elias S. Legal and ethical implications of fetal diagnosis and gene therapy. Am J Med Genet 1990; 35:215-218. [ Links ]
6. Castro I, Mata L. El diagnóstico prenatal de trastornos genéticos. Acta Med Cost 1985; 28:84-91. [ Links ]
7. Castro Volio I, Escalante López G, Mora Palma H, Guerra Carles D, Sánchez Chaves L, Peña Obando C. Cariotipo de células fetales en el diagnóstico prenatal en Costa Rica. Rev Biol Trop 1995; 43 (1-3):31-37. [ Links ]
8. Queenan JT. Amniocentesis, 2a. ed., Nueva Jersey: Medical Economics Books, 1985:201-213. [ Links ]
9. Barch MJ, Lawce HJ, Arsham MS. Peripheral blood culture. En: Barch MJ, ed. The ACT cytogenetics laboratory manual. 2a.ed., Nueva York: Raven, 1991:17-30. [ Links ]
10. Priest JH. Prenatal chromosomal diagnosis and cell culture. En: Barch MJ, ed. The ACT cytogenetics laboratory manual. 2a.ed., Nueva York: Raven, 1991:149-203. [ Links ]
11. Philip J. The prenatal diagnosis and management of congenital malformations in the third trimester of pregnancy. En: Milunsky A, ed. Genetic disorders and the fetus, diagnosis, prevention, and management. 3a. ed., Baltimore: Johns Hopkins Univ. Press, 1992:683-719. [ Links ]
12. Eydoux P, Choiset A, Le Porrier N, et al. Chromosomal prenatal diagnosis: study of 936 cases of intrauterine abnormalities after ultrasound assessment. Prenat diagn 1989; 9:255-268. [ Links ]
13. Wladimiroff JW, Sachs ES, Reuss A, Stewart PA, Pijpers L, Niermeijer MF. Prenatal diagnosis of chromosome abnormalities in the presence of fetal structural defects. Am J Med Genet 1988; 29:289-291. [ Links ]
14. Nicolaides KH, Snidjders RJ, Gosden CM, Berry C, Campbell S. Ultrasonographically detectable markers of fetal chromosomal abnormalities. Lancet 1992; 340:1109-1110. [ Links ]
15. Wilson RD, Chitayat D, McGillivray BC. Fetal ultrasound abnormalities: correlation with fetal karyotype, autopsy findings, and postnatal outcome--five year prospective study. Am J Med Genet 1992; 44:586-590. [ Links ]
16. Twining P, Zuccollo J. The ultrasound markers of chromosomal disease: a retrospective study. Br J Radiol 1993; 66:408-414. [ Links ]
17. Benacerraf BR, Neuberg D, Bromley B, Frigoletto FD. Sonographic scoring index for prenatal detection of chromosomal abnormalities. J Ultrasound Med 1992; 11:449-458. [ Links ]
18. Benacerraf BR, Nadel A, Bromley B. Identification of second-trimester fetuses with autosomal trisomy by use of a sonographic scoring index. Radiology 1994; 193:135-140. [ Links ]
19. Babcook CJ, Goldstein RB, Filly RA. Prenatally detected fetal myelomeningocele: is karyotype analysis warranted?. Radiology 1995; 194:491-494. [ Links ]
20. Nyberg DA, Kramer D, Resta RG et al. Prenatal sonographic findings of trisomy 18: review of 47 cases. J Ultrasound Med 1993; 12:103-113. [ Links ]
21. Nicolaides KH, Campbell S. Ultrasound diagnosis of congenital abnormalities. En: Milunsky A, ed. Genetic disorders and the fetus, diagnosis, prevention and treatment. 3a. ed., Baltimore: The Johns Hopkins Univ. Press, 1992:593-648. [ Links ]
22. Root S, Carey JC. Survival in trisomy 18. Am J Med Genet 1994; 49:170-174. [ Links ]
23. Baty BJ, Blackburn BL, Carey JC. Natural history of trisomy 18 and trisomy 13: I. growth, physical assessment, medical histories, survival, and recurrence risk. Am J Med Genet 1994; 49:175-188. [ Links ]
24. Baty BJ, Jorde LB, Blackburn BL, Carey JC. Natural history of trisomy 18 and trisomy 13: II. psychomotor development. Am J Med Genet 1994; 49:189-194. [ Links ]
25. Faix RG, Barr M, Waterson JR. Triploidy: case report of a liveborn male and an ethical dilemma. Pediatrics 1984; 74:296-299. [ Links ]
26. Romero R, Ghidini A, Santolaya J. Fetal blood sampling. En: Milunsky A, ed. Genetic disorders and the fetus, diagnosis, prevention and treatment. 3a. ed., Baltimore: The Johns Hopkins Univ. Press, 1992:649-682. [ Links ]
* Sección de Genética Humana. Instituto de Investigaciones en Salud (INISA), Universidad de Costa Rica.
** Unidad de Perinatología, Servicio de Obstetricia, Hospital R. A. Calderón Guardia. Caja Costarricense de Seguro Social.
*** Unidad de Perinatología. Servicio de Obstetricia, Hospital México, C. C. S. S.
Correspondencia: Isabel Castro Volio, INISA, Universidad de Costa Rica, San Pedro, Costa Rica

