











 (pdf)
(pdf)




 SciELO
SciELO  SciELO
SciELO 
 Permalink
PermalinkRevista Costarricense de Cardiología
ISSN 1409-4142
Cardiopatías Congénitas en Costa Rica: análisis de 9 años de registro
Dra. Adriana Benavides Laraa, Dra. Lila Umaña Solísb
a. Pediatra, Centro Nacional de Referencia de Enfermedades Congénitas, Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud (INCIENSA). Apartado 4-2250, Tres Ríos, Cartago, Costa Rica. Teléfono: (506)279-9911 (152), Fax: (506)279-5546. benavides@inciensa.sa.cr
b. Pediatra-Genetista, MSC en Epidemiología. Coordinadora Centro Nacional de Registro de Enfermedades Congénitas. Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud (INCIENSA).
Abreviaturas: CC: cardiopatía congénita, CCSS: Caja Costarricense de Seguro Social, CREC: Centro Nacional de Referencia de Enfermedades Congénitas, INCIENSA: Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud, INEC: Instituto Nacional de Estadística y Censos.
Resumen
Introducción: Las cardiopatías congénitas (CC) se encuentran entre las malformaciones congénitas más comunes y tienen gran impacto en la morbilidad y mortalidad pediátricas. El objetivo del presente estudio es explorar la prevalencia y tendencias de las CC en Costa Rica en el período de 1996 al 2004.
Materiales y métodos. El presente es un estudio retrospectivo de base poblacional de los datos del Registro Nacional de Malformaciones Congénitas, anotando variables como año de registro de la CC, tipo, sexo, edad materna y distribución geográfica. Se realizó además un análisis de su impacto en la mortalidad infantil a partir de datos del Instituto Nacional de Estadística y Censos.
Resultados: De todas las malformaciones congénitas las cardiopatías son las más letales y su prevalencia dentro del tiempo estudiado mostró un aumento de 0,10% a 0,18%. Son más frecuentes en varones (p=NS) y la edad materna no demostró ser un factor de riesgo para presentarla. Las CC más frecuentes son los defectos del tabique interventricular. San José y Limón fueron las provincias con mayor prevalencia y Guanacaste con la menor.
Conclusiones: Las CC son las malformaciones más frecuentes y son causa importante de muerte infantil en Costa Rica. Deben mejorarse su diagnóstico perinatal y su registro, con el objetivo de dictar políticas de prevención y atención.
Palabras clave: Cardiopatías congénitas, malformaciones congénitas, mortalidad infantil
Abstract
Introduction: Congenital cardiovascular anomalies constitute one of the largest groups among severe congenital defects and exert an important impact over mortality. This study explored the prevalence and tendencies of congenital cardiovascular defects in Costa Rica between the years of 1996 and 2004.
Methods: A retrospective population based investigation was undertaken from data of the National Registry of Congenital Malformations, taking into account the following factors: type of cardiac anomaly, maternal age, year of registry and geographic distribution of the patients at birth. Besides, we analyzed the impact of a congenital cardiac defect over infant mortality.
Results: The prevalence of congenital heart defects increased from 0,1% to 0,18% during the study period. They exerted greatest impact over infant mortality when compared with any other group of congenital malformations. They show higher frequency in males (p>0.1) and maternal age did not constitute a significant risk factor. The most common lesion detected was ventricular septal defect, finding that agrees with that of other studies from around the world. San José and Limón were the provinces with the highest rates of occurrence and Guanacaste showed the lowest one.
Conclusions: In Costa Rica congenital heart defects have become the malformation with the greatest impact upon infant mortality. This finding represents an important challenge to improve their perinatal diagnosis and their registry with the aim of implementing prevention policies.
Key words: Ventricular septal defect, isolated muscular, perimembranous, septal aneurysm.
Introducción
Se define como cardiopatía congénita (CC) una malformación anatómica del corazón y sus vasos, que ocurre rápidamente en la vida embrionaria desde el día 18, hasta la décimo segunda semana en la vida fetal. Se encuentran entre las malformaciones congénitas más comunes y tienen un gran impacto en la morbilidad y mortalidad pediátricas, así como en los costos de servicios en todas las naciones1-11.
Aunque la prevalencia total de los defectos cardiacos al nacimiento ha sido estimada entre 4 y 9 por 1000 nacimientos2-7,12-15, la cifra exacta depende de la agudeza diagnóstica, los criterios de inclusión de cada registro, factores genéticos y ambientales de cada región, la duración del seguimiento durante el período neonatal de los casos y otros factores concernientes a cada registro. Así, Hoffman y Kaplan en un metanálisis de 62 estudios publicados desde 1955 sobre la incidencia de defectos cardiacos congénitos, determinaron que esta varía entre 4 y 50 por 1000 nacidos vivos, concluyendo que la diferencia en los reportes depende principalmente de la cantidad de defectos cardiacos triviales o menores incluidos, siendo que la incidencia reportada de cardiopatías congénitas moderadas a severas es alrededor de 6 por 1000 nacidos vivos7. Estudios poblacionales recientes en Europa han indicado que el rango de prevalencia varía entre 3,5% 13,7% de los nacidos vivos16.
Además de ser uno de los grupos de defectos congénitos que más vidas cobra alrededor del mundo, presentan el gran reto de que solo el 25% de los nacidos vivos que las padecen son sintomáticos en el período neonatal, y entre ellos varía la edad de su presentación, sean estas mortales o no. En Estados Unidos siguen siendo la causa más común de muerte en la infancia1; un reporte del Registro de Anomalías Congénitas de Glasgow (Reino Unido) indicó que los niños nacidos con CC tienen una sobrevida a los 5 años del 75%, sólo superadas en letalidad por las anomalías cromosómicas, con una sobrevida del 48% y los defectos de tubo neural con un 72%17. Samanek y Vorisková en Bohemia (República Checa) determinaron la sobrevida de los niños nacidos vivos con defectos congénitos a los 15 años en 77,11%18.
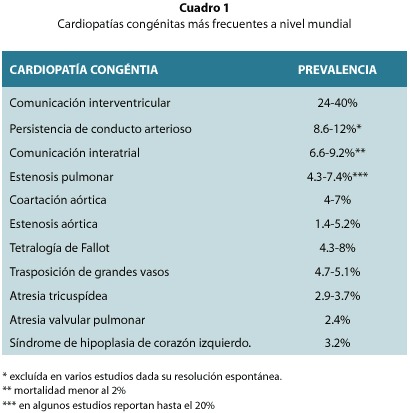
La mayoría de los estudios epidemiológicos publicados es este campo se basan en poblaciones pequeñas, heterogéneas o se refieren a un grupo de defectos muy específicos pero otros han contribuido a dilucidar parte de su epidemiología4,8,12,19 citado en 12, 20. Las definiciones, clasificaciones y sistemas de registro son diferentes entre uno y otro país. Además, pocos defectos específicos son similares para algunas características epidemiológicas, por ejemplo los defectos septales atriales y los ventriculares y los defectos en las almohadillas endocárdicas. Hasta el momento no se ha demostrado en ningún estudio, cambios significativos en la prevalencia de cardiopatías "complejas" o "severas", pero si ha habido un aumento en la prevalencia de defectos menores, principalmente defectos septales, debido probablemente a que se ha agudizado su diagnóstico2,4,5,7,12,20. De hecho, son los más prevalentes en todos los studios (Cuadro 1)6-11-12-19-21-22.
Las causas de CC son complejas e involucran tanto factores genéticos como ambientales; 8% se explican por un defecto genético y existe una recurrencia familiar de 2,3 a 8%, dependiendo del defecto encontrado6. Estudios recientes han identificado genes que juegan un rol importante en la formación cardiaca e inclusive hay "poligenes hipotéticos" que podrían contribuir a malformaciones cardiacas no sindrómicas, que aun están en proceso de identificación9. Otros estudios han determinado anormalidades cromosómicas en hasta 33% de todos los niños muertos por malformaciones cardiacas22 y examinados con autopsia.
En Costa Rica la falta de estudios epidemiológicos en cuanto a gastos médicos, impacto en la mortalidad y posible determinación de factores de riesgo locales de las CC impiden que las políticas de salud puedan orientarse a su prevención y diagnóstico temprano. En la actualidad, no conocemos la prevalencia nacional de malformaciones cardiacas ni su comportamiento a través del tiempo. El objetivo de este estudio es explorar el comportamiento de las malformaciones cardiacas en cuanto a su prevalencia y tendencia, según año, edad materna, sexo y procedencia, además de su impacto en la mortalidad infantil, durante los 7 años que van de 1996 al 2004.
Metodología
El Centro Nacional de Registro de Enfermedades Congénitas (CREC), es un sistema independiente de registro de base poblacional y fuente de averiguación única que proviene de los 24 hospitales con maternidad de la Caja Costarricense de Seguro Social (CCSS) y de 3 hospitales privados, con una cobertura de más del 90% del total de nacimientos del país, en el que se registran todas las malformaciones congénitas detectadas al nacimiento y antes del egreso del recién nacido de la maternidad. Este registro está totalmente integrado al sistema de información de la CCSS y es parte de los informes de declaración obligatoria que se reciben en el Ministerio de Salud, según decreto N° 16488-S publicado en "La Gaceta" de septiembre del 1986.
Este programa se basa en el informe obligatorio de los defectos congénitos, de acuerdo a las normas establecidas. En base a las definiciones, se reportan como malformados el 2% de los nacimientos, esperándose por lo tanto, unos 1 400 reportes anuales según proyecciones.
Se incluyen en el programa todos los nacimientos hospitalarios, vivos o muertos de 500 g o más de peso. No se consideran los nacimientos ocurridos fuera del hospital y que luego ingresan al servicio de neonatología, a menos que dichos nacimientos sean incluidos en el reporte diario del hospital para registro estadístico.
El presente estudio retrospectivo de base poblacional, abarcó alrededor del 90% de los nacimientos nacionales. Como fuente de información se utilizó la base de datos del CREC, de donde se extrajeron todos los reportes de malformaciones cardiacas desde el 1 de enero de 1996 al 31 de diciembre del 2004 y como denominador, la base de datos de nacimientos del Instituto Nacional de Estadística y Censos (INEC). Se estimaron la prevalencia y la tendencia de las CC reportadas al nacimiento durante el período mencionado, tomando en cuenta las variables de año de registro, sexo, edad materna y distribución geográfica. Para estudiar su impacto en la mortalidad, se consignaron las muertes registradas a causa de las CC del registro del INEC durante el período 2001-2004. Para cada prevalencia se determinó un intervalo de confianza del 95%.
Resultados
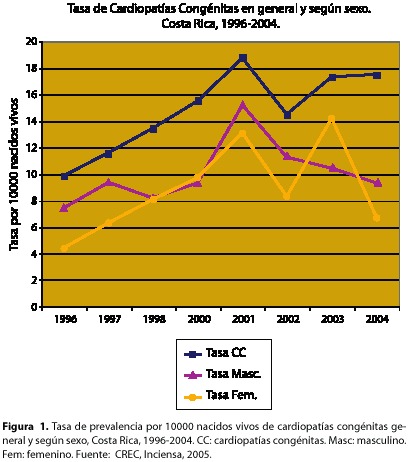
Prevalencia. De 1996 al año 2004 se registraron 1001 malformaciones cardiacas reportadas al nacimiento, es decir, un promedio de 111 por año. La prevalencia de estos defectos congénitos ha aumentado a través de los años de un 0,10% a un 0,18% y aunque es más frecuente en varones, las tasas entre ambos sexos no presentan diferencias significativas, tanto en su magnitud como en su tendencia (figura 1).
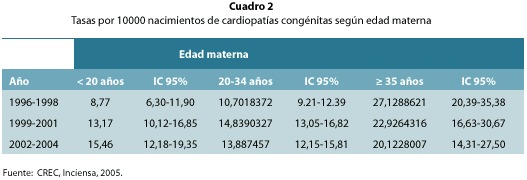
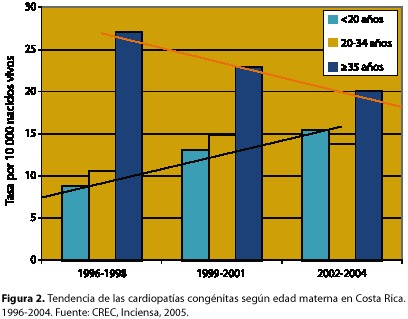
En relación con la edad materna, los nacimientos de madres mayores de 35 años presentaron la mayor tasa de prevalencia de CC, seguidas de los nacimientos de madres menores de 20 años y por último las mujeres entre 20 y 34 años (cuadro 2). Pese a este comportamiento, las diferencias no son estadísticamente significativas. Sin embargo, sí fue significativo el incremento en la tasa de prevalencia de estos defectos en hijos de madres menores de 20 años ya que: aumentó de 8,77 a 15,46 por 10 000 nacidos vivos en los últimos 9 años; sin embargo en los hijos de madres mayores de 35 años, más bien disminuyó (figura 2).
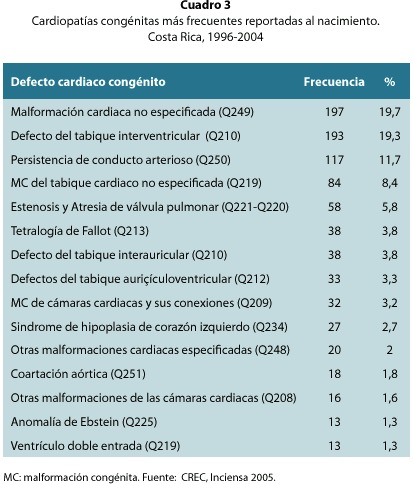
Las CC más frecuentes concuerdan con las reportadas mundialmente, siendo las más prevalentes los defectos de muro interventricular, la persistencia del conducto arterioso, la estenosis y atresia de la válvula pulmonar y la tetralogía de Fallot, así como los defectos del tabique interauricular (cuadro 3). No obstante, nuestro registro está encabezado por las malformaciones cardiacas no especificadas, al igual que en las causas de muerte por cardiopatías del INEC, lo cual marca la gran diferencia con los registros mundiales en cuanto a la capacidad diagnóstica prenatal o durante los primeros días de vida.
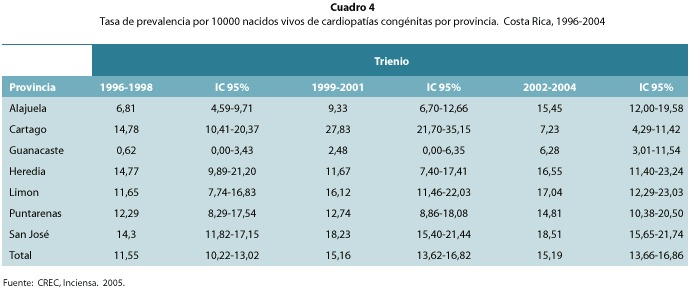
De acuerdo al lugar de procedencia, las provincias con mayor tasa de cardiopatías congénitas en Costa Rica por trienios fueron en su orden San José, Limón, Heredia, Alajuela, Puntarenas, Cartago y Guanacaste (cuadro 4).
Sobre la Mortalidad. Las malformaciones congénitas, principalmente las CC, son la segunda causa de muerte infantil en menores de 1 año después de las causas perinatales y son también la segunda en la población de 1 año a menores de 5, después de las muertes accidentales. La tasa de mortalidad por CC en menores de 1 año es 1,33/ 1000 nacidos vivos. Del total de muertes por CC, 85 % ocurre en el primer año de vida y constituyen el 13% de todas las muertes infantiles (figura 3).
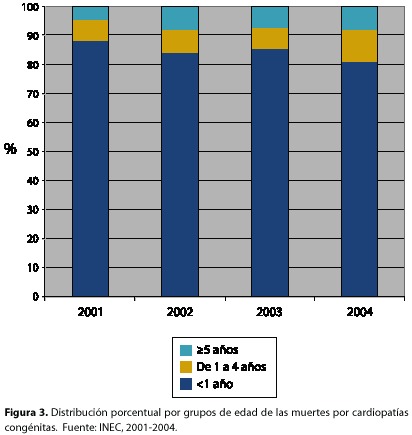
En los niños de 1 a 4 años, el aporte porcentual de las CC en la mortalidad ha venido aumentando con los años (6,5% en el 2004), con un promedio cercano al 40% del total de muertes por malformaciones congénitas.
Analizada según sexo, la tasa de mortalidad infantil por CC en varones es de 1,73/ 1000 nacidos vivos (IC 95% 1,52-1,95) y en mujeres de 1,40/1000 nacidos vivos (IC 95% 1,22-1,61), lo que indica que mueren más varones que mujeres por cardiopatías congénitas, aunque esta diferencia no fue estadísticamente significativa.
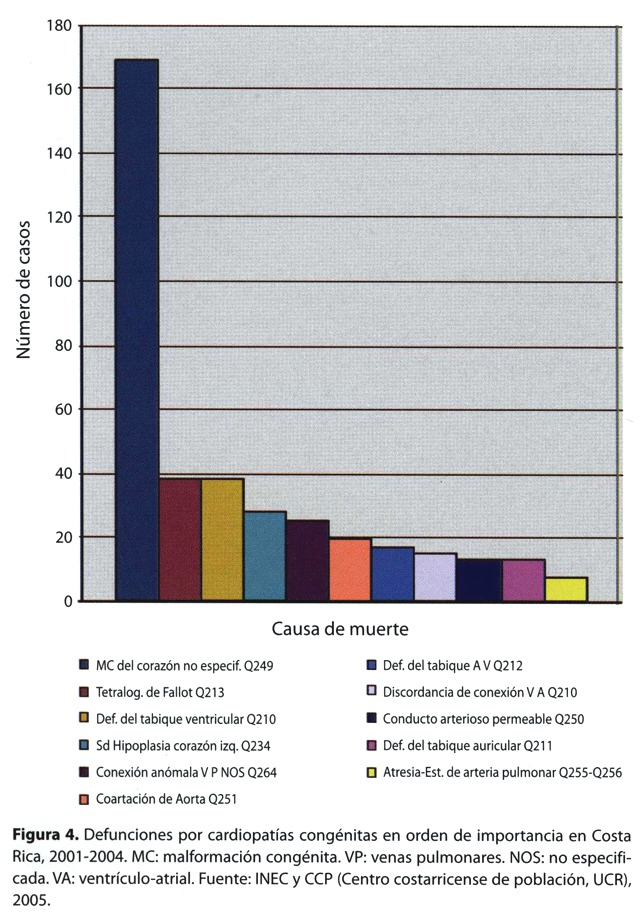
De todas las muertes por CC ocurridas en el período de análisis 2001-2004, el 44% son de causa no especificada, el porcentaje restante muere en su mayoría por tetralogía de Fallot, defectos de tabique ventricular, síndrome de hipoplasia de corazón izquierdo, drenaje anómalo de venas pulmonares, coartación de aorta y los defectos de los tabiques atrio ventriculares (figura 4).
DISCUSIÓN
En Costa Rica los neonatos son egresados de las maternidades al cumplir al menos 12 horas de nacidos, momento en el cual la circulación del niño no se ha adaptado completamente a la vida extrauterina, perdiéndose la oportunidad de diagnosticar y reportar defectos cardiacos que se manifestarán en los siguientes días. Las CC, así como los demás defectos de nacimiento son de reporte obligatorio al CREC antes del egreso de todo recién nacido vivo o muerto de más de 500 g, lo cual excluye todos los abortos y aquellos óbitos que no sean examinados mediante una autopsia. Es por esto que no se tienen datos epidemiológicos fidedignos de los defectos cardiacos congénitos y lo que es peor, que lleguen niños en estado grave a los servicios de emergencia con CC que no fueron diagnosticadas a tiempo.
La tasa de CC ha venido en aumento y en la actualidad se han convertido en el principal defecto congénito grave, con mayor aporte a la mortalidad infantil más que cualquier otra malformación reportada. No se puede afirmar si el aumento es real o si es simplemente un aumento en el reporte o en el diagnóstico de estas anomalías antes de que los niños salgan de las maternidades, pero es posible que aún exista subregistro importante, de ahí que la prevalencia encontrada sea mucho menor a la reportada mundialmente, la cual varía entre 0,7% y 13,7% de los nacimientos2,4-7,12-16. En el Hospital Nacional de Niños (HNN) de la CCSS, centro de referencia nacional de los niños con CC, se diagnostican aproximadamente 640 casos cada año (iincluyen niños de hasta 12 años) y en el CREC, se registran un promedio de 111 CC por año, muchas de las cuales pertenecen a un mismo recién nacido o caso, ya que en el CREC se las registran malformaciones congénitas individualmente. Esta diferencia da una idea del posible subregistro que mencionamos.
En mayo del 2006 se inició un estudio prospectivo, colaborativo INCIENSA-HHNN, con el fin de caracterizar las cardiopatías congénitas en nuestro país y evaluar la calidad del registro. Se esperan resultados para el fin del año 2007, los cuales evaluarán la capacidad de diagnóstico perinatal de CC y de registro obligatorio en niños menores de 1 año y la necesidad de integrar el registro del HNN con el del CREC.
Con respecto a la edad materna, identificada como un factor de riesgo para otros defectos congénitos tales como los defectos de tubo neural y las cromosomopatías, en el presente estudio no demuestra ser un factor de riesgo para las cardiopatías congénitas en general.
Desde el punto de vista de la procedencia, las provincias de Limón y Alajuela han aumentado sus tasas en los últimos 9 años, mientras que en la provincia de Cartago ha disminuido, pese a que antes del año 1998 fue la provincia con mayor prevalencia de este tipo de defectos congénitos (cuadro 5). Por su parte, Guanacaste ha sido, a lo largo del tiempo, la provincia con menor prevalencia de cardiopatías congénitas, siendo esta diferencia con respecto a otras provincias la única estadísticamente significativa. Estas diferencias provinciales son difíciles de explicar dada la complejidad y heterogeneidad de las CC como grupo de malformaciones. No obstante, hipotéticamente varios factores puedan explicar esas diferencias, tales como la adherencia de las diferentes maternidades al registro de casos, las diferencias ambientales entre provincias (por ejemplo Cartago y Limón son provincias, en donde hay mayor uso de pesticidas y tienen en común varios ríos), la diferencia de predominio de razas en las diferentes provincias, la frecuencia de consanguinidad, entre otras. Todas estas variables hipotéticas merecen ser exploradas en un futuro estudio.
Es importante recalcar el subregistro de las causas específicas de muerte en los pacientes con CC. En el HNN la causa no especificada alcanza el 47,2%, lo que quiere decir que en casi la mitad de los niños que mueren en el país por CC no se diagnosticó el defecto cardiaco específico que lo llevó a la muerte8. Parte del problema es que solo a un 35,7% de los niños muertos por esta causa se les realiza autopsia (centro de estadística del mencionado hospital, datos no publicados).
En conclusión, las cardiopatías congénitas se han convertido en el defecto congénito con mayor impacto en la mortalidad infantil y representan un reto muy importante en cuanto a mejorar su diagnóstico perinatal y registro, con el objetivo de dictar políticas de prevención y atención derivadas de estudios epidemiológicos nacionales fidedignos.
RefeRencias
1. Boneva R, Botto L, Moore C, Yang Q, Correa A and Erickson D. Mortality associated with congenital heart defects en the United States: trends and racial disparities, 1979-1997. Circulation 2001; 103: 2376-2381. [ Links ]
2. Wren C, Richmond S and Donalson L. Temporal variability in birth prevalence of cardiovascular malformations. Heart 2000; 83: 414-419. [ Links ]
3. Memberg A, OtterstadJE, Froland G, Hals J and Sorland SJ. Early clinical screening of neonates for congenital heart defects: the cases we miss. Cardiology in the Young 1999; 9: 169-174. [ Links ]
4. Bosi G, Scorrano M, Tosato G, Forini E and Chakrokh R. The Italian Multicentric Study on Epidemiology of Congenital Herat disease: first step of the analysis. Working party of the Italian Society of Pediatric Cardiology. Cardiology in the Young 1999; 9(3): 291-299. [ Links ]
5. Dastgiri S, Stone DH, Le-Ha C and Gilmour WHl. Prevalence and secular trend of congenital anomalies en Glasgow,UK.Archives of Disease Childhood 2002; 86: 257-63. [ Links ]
6. Baekgard Laursen. Some epidemiological aspects of congenital Heart disease in Denmark. Acta Paediatric Scandinavian 1980; 69: 619-624. [ Links ]
7. Hoffman JI, Kaplan S. The incidence of congenital heart disease. Journal of American College of Cardiology 2002; 39(12): 1890-1900 [ Links ]
8. Instituto Nacional de Estadística y Censos (base de datos en internet). Instituto Nacional de Estadística y Censos de Costa Rica, INEC. Disponible en: http://ww.inec.go.cr (accesado el 5 de febrero del 2006). [ Links ]
9. Brennan P and Young ID. Congenital heart malformations: aetiology and associations. Seminars in Neonatology 2001; 6: 17-25. [ Links ]
10.Kuehl K, Loffredo C and Ferencz Ch. Failure to diagnose congenital heart disease in infancy. Pediatrics 1999; 103:743-748. [ Links ]
11.McMillan J, DeAngelis C, Feigin R and Warshaw J. Oski s Pediatrics: Principles and practice. Third Edition. Lippincott Williams and Wilkins, Philadelphia. 1999; 1322-1376. [ Links ]
12. Pradat P, Francannet C, Harris JA and Robert E. The Epidemiology of Cardiovascular Defects, Part 1: a study based on data from three large registries of congenital malformations. Pediatr Cardiol. 2003; 24: 195-221. [ Links ]
13. Harris JA, Francannet C, Pradat P and Robert E. The Epidemiology of Cardiovascular Defects, Part 2: a study based on data from three large registries of congenital malformations. Pediatr Cardiol. 2003; 24: 222-235. [ Links ]
14.Carlgren LE, Ericson A, Kallen B. Monitoring of congenital cardiac defects. Pediatr Cardiol 1987; 8: 247-256. [ Links ]
15. Hanna EJ, Nevin NC, Nelson J. Genetic study of congenital heart defects in Northern Ireland 1974-1978. J Med Genet 1994; 31: 858-863. [ Links ]
16. Hoffman JIE. Incidence, mortality and natural history. En: Anderson RH, Macartney FJ, Shineborne, et al, editors. Pediatric Cardiology. London: Churchil Livingstone; 2002: 111-139. [ Links ]
17.Dastgiri S, Gilmour WH and Stone DH. Survival of children born with congenital anomalies. Arch Dis Child. 2003; 88: 391-394. [ Links ]
18. Samanek M.,Voriskova M. Congenital heart diseases among 815569 children born between 1980 and 1990 and their 15 year survival: a prospective Bohemia survival study. Pediatric Cardiology 1999; 20: 411- 417. [ Links ]
19. Ferencz C, Correa-Villaseñor A, Loffredo CA, Wilson PD. Genetics and enviromental risk factors of major cardiovascular malformations:The Baltimore-Washington Infant Study: 1981-1989. En: Perspectives in Pediatric Cardiology Vol. 5, Futura Publishing, Armonk, NY; 1997. [ Links ]
20. Bosi G, Garani G, Scorrano M, Calzolari E and The IMER Working Party. Temporal variability in birth prevalence of congenital heart defects as recorded by a general birth defects registry. J Pediatr. 2003; 142: 690- 698. [ Links ]
21. Pexieder T, Bhoch D, EUROCAT Working Party on Congenital Heart Disease. EUROCAT Subproject on epidemiology of congenital heart diseases: first analisys of the completed study. En: Clark EB, Markwald RR, Takao A, editors. Developmental mechanism of heart disease. Armonk, NY: Futura Publishing Co; 1995: 655-678. [ Links ]
22.Tennstedt C, Chaoui R, Corner H and Dietel M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: results of seven year necropsy study. Heart 1999; 82(11): 34-39. [ Links ]
23. Organización Panamericana de la Salud.Clasificación Estadística Internacional de Enfermedades y Problemas Relacionados de Salud. Décima Revisión. Volumen 1. Washington DC: OPS, 1995. [ Links ]

