











 (pdf)
(pdf)




 SciELO
SciELO  SciELO
SciELO 
 Permalink
PermalinkRevista Costarricense de Cardiología
ISSN 1409-4142
Síndrome metabólico: ¿Un elefante en una caja?
Dr. José Luis Quirós Alpízara, Bach. Lisa Miranda Solísb, Dr. Juan Pablo Solís Barqueroc
a Médico residente de patología, Hospital San Juan de Dios, San José, Costa Rica
b Bachiller en ciencias médicas, Universidad de Costa Rica
c Médico residente de medicina nterna, Hospital San Juan de Dios, San José, Costa Rica
Resumen
El síndrome metabólico (SM) es una entidad compleja, que incluye factores de riesgo predictores de enfermedad cardiovascular. Entre ellos podemos mencionar la obesidad, diabetes e hipertensión arterial, pero sus criterios y definición en el síndrome metabólico han sido revisadas y criticadas en diversas publicaciones médicas. Presentamos una revisión de la fisiopatología del SM, destacando la participación del tejido adiposo en la resistencia a la insulina, y las guías terapéuticas de la Federación Internacional de Diabetes.
Palabras clave: sindrome metabólico, resistencia insulina, enfermedad cardiovascular, diabetes mellitus, hipertensión arterial, obesidad.
Abstract
The metabolic syndrome (MS) is a complex entity that includes a group of risk factors used in the prediction of coronary disease. These factors include obesity, diabetes and hypertension, but the syndrome has been criticized and revised in various medical journals. We present a review of the physiopathology of the MS, especially of the role of the adipose tissue in insulin resistance, and the new international Federation of Diabetes treatment guidelines for MS.
Key words: metabolic syndrome, insuline resistence, cardiovascular disease, diabetes mellitas, hipertensión, obesity.
Introducción
En la sesión anatomopatológica No. 1663 del 18 de marzo del 2005 en el Hospital San Juan de Dios, en Costa Rica, se presentó el caso de una mujer de 40 años con enfermedad coronaria severa1. Uno de los aspectos discutidos en ella fue la trascendencia que podría haber tenido para esa paciente el diagnóstico temprano de síndrome metabólico. Sin embargo, desde esa fecha a la actualidad, se han suscitado tantos cambios en la forma de entender a esta entidad, que si la discusión fuese hoy, probablemente nuestros conceptos serían diferentes a los de hace un año. Y es que precisamente este ha sido uno de los temas que más radicalmente ha evolucionado en el quehacer médico, con miles de artículos que destacaron su importancia y luego muchos otros que la rechazaron. En este sentido, el título de un artículo editorial describe muy bien el problema: "Síndrome metabólico. Una nueva definición o una gran controversia"2. El final de aquella sesión concluiría con un mensaje muy oportuno: la moderadora mostró una imagen tomada de un texto de electrocardiografía "The blind men and the elephant"3 mostrando a varios hombres tratando de visualizar al enorme animal a través de agujeros muy pequeños, Uno de ellos decía: "es un árbol, pues veo un tronco", otro decía "es una espada" al tocar un colmillo, y otro, "es una serpiente", al tocar la cola. No lograron ponerse de acuerdo porque cada uno llegó a una verdad parcial, pues no consideraron el problema de manera integral, tal como podría estar ocurriendo con nuestra visión del síndrome metabólico.
Historia
Ya en 1923, un investigador de apellido Kylin describió la asociación de hipertensión arterial (HTA), hiperglicemia y gota como un síndrome4. Posteriormente, en 1983 se describen por primera vez los factores de riesgo ateroescleróticos agrupados5. Para el año 1988, Gerald Reaven describe el síndrome X, usando este término para referirse a la asociación de dislipidemia, HTA, enfermedad arterial coronaria y resistencia a la insulina6. Es importante recalcar que existe otra entidad denominada síndrome X cardíaco, caracterizada por la presencia de angina de pecho con angiografía coronaria normal, que se ha correlacionado con síndrome metabólico, pero que difiere del síndrome X descrito por Reaven.6,7
Finalmente en 1998, la Organización Mundial de la Salud (OMS) propuso una nueva definición para este síndrome y lo llamó síndrome metabólico (SM), en lugar de síndrome de resistencia a la insulina4. Desde entonces se lo define como la agrupación de factores de riesgo metabólicos cuyo origen se relaciona con la obesidad, el sedentarismo y factores genéticos que tienen en común la resistencia a la insulina y que predicen la aparición de enfermedad cardiovascular y diabetes mellitus tipo 2 (DM 2).8
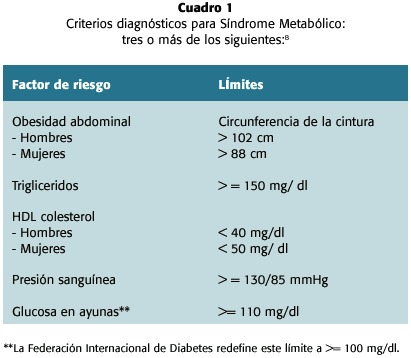
Subsecuentemente, otras anormalidades metabólicas diversas han sido asociadas con este síndrome, las que incluyen la microalbuminuria y las alteraciones en la fibrinólisis y la coagulación, redefiniéndola como una condición inflamatoria sistémica asociada a un estado protrombótico9. En el 2001, en el Programa Nacional de Educación sobre el Colesterol (National Cholesterol Education Program- NECP), el III Grupo de Expertos para su Tratamiento en Adultos (Adult Treatment Panel III, ATP-III) puntualizó la importancia del SM y estableció criterios sencillos que permitan al médico realizar el diagnóstico8 (cuadro 1). Este síndrome se encuentra en un constante proceso de redefinición, por ejemplo, recientemente la Federación Internacional de Diabetes ha modificado uno de sus criterios diagnósticos2.
Otros términos con que se ha referido la literatura científica a este síndrome incluyen: síndrome metabólico múltiple, síndrome de resistencia a la insulina, cuarteto mortal, síndrome DROP (Dyslipidemia, insulin Resistence, Obesity, high blood Pressure ), dislipidemia metabólica y síndrome plurimetabólico4,10,11.
Fisiopatología
Diversos estudios epidemiológicos han demostrado un aumento en la incidencia de este síndrome4. Su prevalencia en los Estados Unidos desde 1988 hasta 1994 fue estimada en un 23,7%12,13,14. En nuestro país se han publicado 2 estudios que abarcaron este tema. En el año 1997 se publicó un trabajo que analizó a 100 pacientes de la consulta externa de endocrinología del Hospital Calderón Guardia, analizados desde el punto de vista clínico y metabólico, encontrándose una fuerte asociación entre hiperinsulinemia, obesidad y acantosis nigricans14. Posteriormente Alvarado y Jiménez realizaron un estudio retrospectivo entre el 2001 y el 2002 en el nivel de atención primaria de Nicoya, en pacientes diabéticos e intolerantes a los carbohidratos, encontrando que en esta población el 68.6% cumplía con los criterios diagnósticos del síndrome metabólico15.
Resistencia a la insulina (RI). El SM está estrechamente relacionado a un desorden metabólico conocido como RI, en el cual la respuesta de los tejidos a la acción normal de esta hormona está alterada5,16,17,11. La mayoría de las personas con RI tienen obesidad abdominal y a pesar de que los mecanismos que conectan a estos dos elementos son complejos y "desconocidos", muchas publicaciones sugieren una función endocrina del tejido adiposo19,20. Anteriormente se pensaba que el tejido adiposo era sólo un depósito pasivo de almacenamiento del exceso de calorías. Sin embargo, hay evidencia reciente que indica que los adipocitos sintetizan y secretan moléculas biológicamente activas convirtiendo al tejido adiposo en un órgano endocrino. Ellas incluyen leptina, resistina, adiponectina, factor de necrosis tumoral alfa (TNF-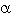 ), interleucina 6 (IL-6), inhibidor del activador del plasminógeno de tipo 1 (PAI-1), ácidos grasos y angiotensinógeno21.
), interleucina 6 (IL-6), inhibidor del activador del plasminógeno de tipo 1 (PAI-1), ácidos grasos y angiotensinógeno21.
El eje central del síndrome es un fenotipo de obesidad-hiperin- sulinemia-dislipidemia, el cual predice el desarrollo incidente de DM 2 y de enfermedad cerebro vascular22,23,24.
La insulina ejerce sus acciones intracelulares por medio de dos vías de señalización: la vía del fosfatidil-inositol-3-quinasa (PI-3K) y la vía de la proteínquinasa activada por mitógenos24 (MAPK, mitogen-activated protein kinase). Las acciones anabólicas ejercidas por la insulina a través de la vía PI-3K permiten un incremento en el transporte y utilización de la glucosa, disminución de la lipólisis en el tejido adiposo y un aumento en la liberación de óxido nítrico (NO). Todas estas acciones tienen efecto antiaterogénico. A través de la MAPK se promueven acciones celulares relacionadas con el crecimiento y la mitosis23,24,25.
Se ha postulado que la RI aparece en la vía de la PI-3K, resultando en un estímulo aumentado de la vía MAPK, la cual conlleva a la proliferación de las células del músculo liso vascular y aumento de la síntesis de las proteínas de la matriz extracelular en la pared de los vasos sanguíneos24. Sin embargo, el estímulo mitógeno de la insulina sobre las células del músculo liso vascular es débil y podría no ser tan significativo23; por lo tanto, se ha propuesto que las acciones aterogénicas de la insulina se ejecutan por intermedio de otras sustancias más potentes como por ejemplo, el factor de crecimiento derivado de plaquetas26.
La insulina, cuya liberación es mediada por la ingesta de alimentos, produce un aumento en la actividad del sistema simpático. Por efecto de la insulina, la glucosa es internalizada en las células del núcleo ventromedial del hipotálamo, lo que produce una supresión de vías inhibitorias entre las células reguladoras de la corteza cerebral y las de los centros reguladores del sistema simpático del tallo cerebral lo que, consecuentemente, aumenta la actividad simpática19. Hay evidencia adicional que sugiere participación del sistema simpático en la relación entre insulina e HTA.
La RI y la hiperinsulinemia son más severas y más fuertemente relacionadas con la HTA en personas obesas que en no obesas. De acuerdo a esto, la hiperinsulinemia –una consecuencia de la RI-, estimula al sistema simpático, aumentando la termogénesis. Sin embargo, el aumento en la actividad simpática contribuye a la HTA, al estimular al miocardio, al sistema vascular y al riñón. Por lo tanto, la HTA relacionada a la obesidad puede ser una consecuencia no deseada de los mecanismos fisiológicos que reestablecen el balance energético y estabilizan el peso corporal19.
En un estudio de cohortes derivado del Normative Aging Study en Boston, la actividad simpática, medida por la excreción urinaria de norepinefrina en orina de 24 h y corregida según el índice de masa corporal, estuvo elevada en sujetos con hiperinsulinemia. Además, los sujetos obesos no tuvieron resistencia a la acción de la insulina sobre el sistema simpático, evidenciado por los aumentos en la concentración de norepinefrina plasmática, a pesar de una disminución marcada en la captación de glucosa estimulada por la insulina19. Estos hallazgos son consistentes con la hipótesis de que la estimulación simpática en pacientes obesos es mediada por la insulina.
Alternativamente, la asociación entre RI e HTA podría resultar de un aumento primario en la actividad del sistema simpático. El aumento en la actividad simpática refleja, antagoniza la captación de glucosa mediada por la insulina. Sin embargo, a pesar de que un aumento primario en la actividad simpática podría causar tanto HTA como RI, estudios en animales indican que la RI inducida por dieta rica en grasa ocurre antes de la aparición de HTA19, aunque es otra observación consistente con la estimulación del sistema simpático mediada por la insulina. Los estudios con somatostatina también aportan evidencia adicional acerca del estímulo simpático mediado por insulina. Esta hormona que inhibe la secreción endógena de insulina, disminuye los niveles de norepinefrina y disminuye la presión arterial en pacientes con hiperinsulinemia. Sin embargo, la supresión farmacológica del sistema simpático con guanadrel (un antagonista adrenérgico), no redujo la RI en un estudio con pacientes hipertensos19.
En pacientes no obesos con HTA, se desconoce la causa de la RI. Las intervenciones no farmacológicas, como la restricción calórica, la pérdida de peso y el ejercicio, las cuales producen disminución de la RI, disminuyen también la actividad simpática y la presión arterial19. Dado que la insulina es un vasodilatador directo, son necesarias otras hipótesis fisiopatológicas que establezcan un rol de la insulina en la patogénesis de la HTA. El sistema simpático, así como otros componentes indefinidos del estado de RI, pueden antagonizar el efecto vasodilatador normal de la insulina en pacientes obesos y en aquellos con HTA19. Asimismo, los pacientes con insulinomas no tienen mayor prevalencia de HTA que aquellos sin insulinotas; en estos pacientes, la hiperinsulinemia es primaria, no debida a RI19.
Dislipidemia. El incremento de los niveles de catecolaminas puede explicar el aumento de la síntesis de lipoproteínas de muy baja densidad (VLDL), lo cual conlleva al aumento de la concentración plasmática de triglicéridos y la disminución de la síntesis de lipoproteínas de alta densidad (HDL)8,10. Así, la relación triglicéridos/HDL colesterol, es un poderoso predictor de riesgo tanto de resistencia a la insulina como de enfermedad cardiovascular8,10.
Inflamación y estado pro-trombótico. Se ha documentado en diversos estudios que la hiperinsulinemia predice la incidencia de enfermedad cardiovascular, la cual se asocia a una condición inflamatoria sistémica y a un estado pro-trombótico2,21. Diferentes estudios han sugerido el concepto de un estado pro-inflamatorio como parte del SM. Así, se ha observado relación entre la proteína C reactiva (PCR) como factor predictor de enfermedad coronaria. Se ha propuesto que el aumento en la liberación de diversas citoquinas por parte del tejido adiposo, conducen a un aumento en la producción de PCR por parte del hígado.27
El estado pro-trombótico21 es favorecido por niveles elevados de PAI-1, factor de von Willebrand (vWF), fibrinógeno y por la disfunción endotelial28. La actividad del PAI-1 (un inhibidor de la fibrinólisis), está aumentada en hombres jóvenes que sobrevivieron a un infarto agudo del miocardio y predice la recurrencia de eventos trombóticos24,29. Además, los pacientes con DM 2 presentan niveles aumentados de PAI-1, particularmente quienes presentan enfermedad cardiovascular24(fig. 1).
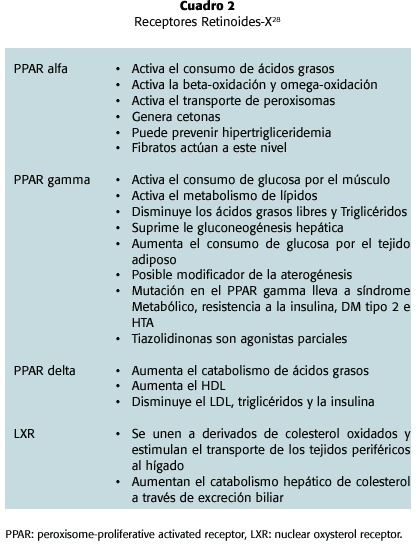
Otro enfoque innovador para intentar comprender a este elefante es a través de la familia de receptores intracelulares conocidos como retinoides-X. Estos son heterodímeros que se unen a una variedad de ligandos derivados de los ácidos grasos y el colesterol y que son reguladores de genes diana que median el transporte y catálisis de los lípidos; algunos de ellos se han relacionado, unos con más importancia que otros, en la fisiopatología del SM (cuadro 2)28. El estudio de estos receptores y de este ciclo da nuevas bases moleculares y fisiológicas para el estudio y experimentación de nuevos medicamentos para el tratamiento del SM.
Tratamiento
El manejo del SM, que debe verse en esta entidad como un conjunto30,31, se enfoca en la detección temprana de los factores de riesgo, y el manejo oportuno de cada uno de ellos. Se deben identificar los factores genéticos y ambientales que propicien este síndrome e insistirse en el manejo no farmacológico y la modificación de los estilos de vida perjudiciales32,33. Sin embargo, hay aún controversia en cuanto a su definición. El último consenso de la Asociación Americana de Diabetes y la Asociación Europea para el estudio de la Diabetes establece una visión crítica sobre la definición del SM34,35. Una de las críticas más importantes se refiere a la estandarización de los criterios diagnósticos, ya que las definiciones planteadas por el ATP III y la OMS difieren o algunas de ellas no se establecen claramente, como por ejemplo, en la definición de HTA, en la que no se especifica si las cifras de presión diastólica y sistólica propuestas para diagnosticar SM son diagnósticas por separado o si se requiere la elevación de ambas; tampoco se especifica si un paciente previamente hipertenso que se encuentre compensado cumple con el criterio para diagnosticar SM34,35. También se cuestiona si el SM per se es predictor de enfermedad cardiovascular, ya que si bien la mayoría de los estudios ha mostrado que los pacientes que cumplen los criterios de SM tienen una mayor prevalencia de enfermedad cardiovascular, la falta de una metodología estandarizada para alcanzar la definición del ATP III disminuye su valor predictivo. No hay estudios en donde se incluya otros factores de riesgo convencionales para valorar la especificidad del SM. Aún no queda claro si etiquetar a cientos de pacientes con el diagnóstico de SM esté logrando un claro beneficio en la práctica clínica34. Muchos investigadores que han dedicado su vida a su investigación cuestionan su utilidad, entre ellos Reaven, quien plantea que ha tomado más importancia su clasificación que el verdadero propósito que se tenía a la hora de definir un síndrome, cual es la prevención de enfermedad cardiovascular6,34,35,36. Finalmente, son aleccionadoras las palabras de Margo A. Denke: " el síndrome metabólico se parece a un elefante y todas las revisiones de su importancia se encuentran ignominiosamente reducidas al examen de los colmillos, trompas y cola nuestra sociedad ha desviado su mirada de las recompensas a largo plazo que dan las buenas conductas sostenibles. Con el fin de domar las conductas que estimulan el SM, aquellas preguntas sencillas surgidas de la dieta y el tratamiento farmacológico necesitan el apoyo de la sociedad para ser eficaces"30.
Referencias
1. Quirós JL, Miranda L, Celada VE. Síndrome metabólico en relación a poliquistosis ovárica y mutación MTHFR: Caso Clínico. Gaceta Médica de Costa Rica 2005; 7: 79-83. [ Links ]
2. Quesada O. Síndrome metabólico: Una nueva definición o una gran controversia. Actualización Médica Periódica (Costa Rica) 2005; 54: 1-11 [ Links ]
3. Blake TM. The blind men and the elephant. En: Introduction to electrocardiography. Nueva York: Meredith Corporation, 1964; 17. [ Links ]
4. Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care 2001; 24: 683-689. [ Links ]
5. Wingard DL, Barrett-Connor E, Criqui M, Suarez L. Clustering of heart disease risk factors in diabetic compared to nondiabetic adults. Am J Epidemiol 1983; 117: 19-26. [ Links ]
6. Reaven GM. Banting lecture 1988: role of insulin resistance in human disease. Diabetes 1988; 37: 1595-1607. [ Links ]
7. Monti LD, Piatti PM. Role of endothelial dysfunction and insulin resistance in angina pectoris and normal coronary angiogram. Heart 2005; 30: 48-54. [ Links ]
8. Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults. Executive summary of the Third report of the National Cholesterol Education Program (NCEP) expert Panel on Detection, evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285: 2486-2497. [ Links ]
9. Yudkin JS. Abnormalities of coagulation and fibrinolysis in insulin resistance. Diabetes Care 1999; 22: C25-C30. [ Links ]
10. Scott CL. Diagnosis, prevention, and intervention for the metabolic syndrome. Am J Cardiol 2003; 92: 35i-42i. [ Links ]
11. Denke MA. Síndrome metabólico. Current Atherosclerosis Reports (edición en español) 2003; 1: 72-75. [ Links ]
12. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among Us adults: findings from the third National Health and nutrition examination survey. JAMA 2002; 287: 356-359. [ Links ]
13. Ford ES, Giles WH. A comparison of the prevalence of the metabolic syndrome using two proposed definitions. Diabetes Care 2003; 26: 575-581. [ Links ]
14. Fernández E, Morera O. Síndrome X en Costa Rica (diabetes mellitus tipo II, obesidad con estigma dérmico). Rev Med de Costa Rica y Centroamérica 1997; 538: 35-38. [ Links ]
15. Alvarado V, Jiménez MF. Síndrome metabólico en pacientes diabéticos tipo 2 e intolerantes a carbohidratos del EBAIS La Mansión, Nicoya. Acta Med Costarric 2003; 45: 154-17. [ Links ]
16. Hsueh WA, Quinones MJ. Role of endothelial dysfunction in insulin resistance. Am J Cardiol 2003; 92:10J-17J. [ Links ]
17. Reaven GM. Metabolic syndrome: pathophysiology and implications for management of cardiovascular disease. Circulation 2002; 106: 286-288. [ Links ]
18. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities--the role of insulin resistance and the sympathoadrenal system. N Engl J Med 1996; 334: 374-381. [ Links ]
19. Sowers JR. Obesity as a cardiovascular risk factor. Am J Med 2003; 115: 37S-41S. [ Links ]
20. Reilly MP, Rader DJ. The metabolic syndrome: more than the sum of its parts? Circulation 2003; 108: 1546-1551 [ Links ]
21. Shuldiner AR, Yang R, Gong D. Resistin, obesity, and insulin resistance -the emerging role of the adipocyte as an endocrine organ. N Engl J Med 2001; 345: 1345-1346. [ Links ]
22. Deedwania PC. The deadly quartet revisited. Am J Med 1998; 105: 1S-3S [ Links ]
23. King GL, Wakasaki H. Theoretical mechanisms by which hyperglycemia and insulin resistance could cause cardiovascular diseases in diabetes. Diabetes Care 1999; 22: C31-C37. [ Links ]
24. Fagan TC, Deedwania PC. The cardiovascular dysmetabolic syndrome. Am J Med 1998; 105: 78S-82S. [ Links ]
25. Meigs JB, Wilson PW, Nathan DM, DAgostino RB, Williams K, Haffner SM. Prevalence and characteristics of the metabolic syndrome in the san Antonio Heart and Framingham Offspring studies. Diabetes 2003; 52: 2160-2167. [ Links ]
26. Graf K, Xi XP, Yang D, Fleck E, Hsueh WA, Law RE. Mitogen-activated protein kinase activation is involved in platelet-derived growth factor- directed migration by vascular smooth muscle cells. Hypertension 1997; 29: 334-339 [ Links ]
27. Grundy SM. Inflammation, metabolic syndrome, and diet responsiveness. Circulation. 2003; 108:126-128. [ Links ]
28. Shulman AI, Mangelsdorf DJ. Retinoid X receptor heterodimers in the metabolic syndrome. N Engl J Med 2005; 353: 604-615. [ Links ]
29. Hamsten A, de Faire U, Walldius G, Dahlen G, Szamosi A, Landou C et al. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet 1987; 2: 3-9 [ Links ]
30. Denke MA. Metabolic syndrome. Curr Atheroscler Rep 2002; 4: 444- 447. [ Links ]
31. Scott CL. Diagnosis, Prevention, and intervention for the Metabolic syndrome. Am J Cardiol 2003; 92: 35i-42i. [ Links ]
32. Grundy SM, Hansen B, Smith SC Jr, Cleeman JI, Kahn RA et al. Clinical Management of Metabolic syndrome: report of the American Heart Association/National Heart, Lung, and Blood Institute/American Diabetes Association conference on scientific issues related to management. Circulation 2004; 109: 551-556 [ Links ]
33. Wilson PW, Grundy SM. The Metabolic syndrome: Practical Guide to Origins and Treatment: Part I & II. Circulation. 2003; 108: 1422-1424 y 1537-1540 [ Links ]
34. Kahn R, Buse J, Ferrannini R, Stern M. The Metabolic syndrome: time for a critical appraisal. Joint statement from the American Diabetes Association for the study of Diabetes. Diabetes Care 2005; 28: 2289- 2304. [ Links ]
35. Zimmet P, Alberti GMM, Serrano M. Una nueva definición mundial del syndrome metabólico propuesta por la Federación Internacional de Diabetes: fundamento y resultados. Rev Esp Cardiol 2005; 58: 1371- 1376. [ Links ]
36. Reaven GM. The Metabolic syndrome: requiescat in Pace. Clin Chem 2005; 51: 931-938. [ Links ]
a. Autor para correspondencia: Servicio de patología, Hospital San Juan de Dios, San José, Costa Rica. Teléfono 533-1291. Fax 256-0153. E-mail quiros@yahoo.com

